Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Cognitive impairment after surgery is a common problem, affects mainly the elderly, and can be divided into postoperative delirium and postoperative cognitive dysfunction. Both phenomena are accompanied by neuroinflammation; however, the precise molecular mechanisms underlying cognitive impairment after anesthesia are not yet fully understood. Anesthesiological drugs can have a longer-term influence on protein transcription, thus, epigenetics is a possible mechanism that impacts on cognitive function. Epigenetic mechanisms may be responsible for long-lasting effects and may implicate novel therapeutic approaches.
- epigenetics
- anesthesia
- inflammation
1. Molecular Mechanisms of Postoperative Cognitive Impairment
The pathophysiology of POD and POCD is closely related to neuroinflammation [23]. The biomarkers currently discussed display mainly a set of not only inflammatory markers [24], among them interleukin (IL-)6, C-reactive protein [25], IL-8, IL-10 and matrix metalloproteinase 8 [26], but also the brain-related markers S100 calcium-binding protein B and neuron-specific enolase [15]. In addition, markers belonging to the cholinergic system, such as acetylcholinesterase and butyrylcholinesterase, have been studied and assessed as possible biomarkers for delirium [27].
The current understanding of the development of POD and POCD is that surgery causes trauma of the surgical site accompanied by the release of damage-associated patterns [23,28] (Figure 1). The most prominent damage-associated patterns in this context are high mobility group protein B1 (HMGB1) and S100 calcium-binding protein A8 [29]. HMGB1 can bind to toll-like receptors TLR2 and TLR4 of different immune cells, and activates nuclear factor kappa B, which, in turn, causes the release of several cytokines [30]. The pro-inflammatory cytokines IL-1 beta (IL-1β), IL-6 and tumor necrosis factor alpha cause a further release of HMGB1 in a positive feedback loop, amplifying the inflammatory response and the upregulation of cyclooxygenase 2 isozyme release [31]. Circulating peripheral pro-inflammatory cytokines disrupt blood-brain barrier (BBB) permeability via cyclooxygenase 2 upregulation and MMPs, allowing pro-inflammatory cytokines to enter the central nervous system (CNS) [32]. After the disruption of the BBB, peripheral macrophages can enter the brain, triggered by monocyte chemoattractant protein 1, and can activate microglia, which are known as the resident macrophages of the CNS [33]. When microglia are activated, they themselves release cytokines, and neuroinflammation is amplified. In addition, microglia can release reactive oxygen species in response to HMGB1, which aggravates cognitive impairment [31,34]. Disruption of the BBB, in turn, leads to the release of CNS-specific proteins S100 calcium-binding protein B and neuron-specific enolase to the plasma [35] (Figure 1). In addition, microglia astrocytes are activated and promote inflammation and the release of glutamate and glycogen [17]. All these processes cause damage to several functions in the brain, including the depression of inhibitory neurotransmission via downregulation of gamma-aminobutyric acid receptors, disrupting the balance of excitatory and inhibitory neurotransmission and, ultimately, favoring glutamate toxicity [36], the pathological activation of astrocytes, which causes synaptic dysfunction [28], and the reduction of cerebral acetylcholine, triggering the complex neuroinflammatory response and the subsequent degeneration of cholinergic neurons through neural necrocytosis, apoptosis and pyroptosis [13]. In addition, the cholinergic anti-inflammatory pathway seems to play an important role in the pathophysiology. Surgical trauma induces activation of the sympathetic nervous system, and the vagus reflex is stimulated, which causes the release of acetylcholine and activates the nicotinic α7 receptor on macrophages [28], which causes the inactivation of the nuclear factor ‘kappa-light-chain-enhancer’ of activated B (NF-kB) cells and decreases cytokine release [37,38]. Hence, vagus nerve stimulation may attenuate postoperative cognitive impairment, but a reduced secretion of acetylcholine could aggravate inflammation [39], and acetylcholinesterase inhibitors have been discussed as potential treatment options for cognitive impairment [40,41]. As the effects caused by surgical trauma are relatively well described, it still remains elusive how anesthetics impact on cognitive impairment.
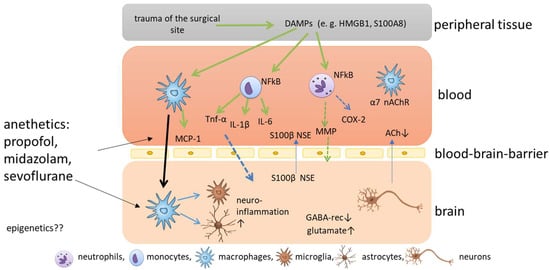
Figure 1. Schematic overview of the molecular mechanisms of postoperative cognitive impairment. Trauma of the surgical site causes the release of damage associated patterns (DAMPS), which are released into the blood where nuclear factor kappa B is activated in immune cells, such as neutrophils, monocytes and macrophages, which release cytokines and chemokines. The release of matrix metalloproteinases (MMPs) and cyclooxygenase 2 (COX2) cause permeabilization of the blood-brain barrier and pro-inflammatory cytokines and macrophages enter the brain. Here, microglia are activated and neuroinflammation is amplified.
2. Epigenetic Mechanisms
It has been proposed earlier that epigenetics might contribute to cognitive impairment for several reasons. However, until 2013, no heritable, genomic indices of persistent POCD or postoperative dementia after surgery had been identified [6].
The risk factors identified as contributing to cognitive impairment are, inter alia, exposure to general anesthesia, hypotension, hypoxia, psychoactive drugs and hippocampal inflammation induced by the surgical intervention. In addition, earlier studies indicate that these factors might induce epigenetic dysfunction in the brain, as chromatin remodeling is required for memory-associated gene transcription and expression [19]. Additional epigenetic and environmental factors are supposed to accompany increasing age and play a significant role in the pathogenesis postoperative cognitive impairment, which mainly occurs in postoperative elderly patients and is characterized by troubles in cognition [53].
Epigenetics Mechanisms Induced by Anesthetics
It has recently been suggested that unknown, novel, epigenetic-related mechanisms could cause anesthetic-induced neuronal toxicity to human neurons, and that epigenetic modifiers, such as DNA-methyltransferase (DNMT) and histone deacetylase inhibitors (HDACis), might be promising targeted therapeutics to mitigate the neurotoxic effects of anesthetics in the developing brain [54].
However, little is known about whether anesthetics induce epigenetic changes in genes related to cognitive impairment, and further research is needed to elucidate the epigenetic mechanisms completely and their role in this field.
Epigenetic modification generally includes variations in DNA methylation, histone acetylation, histone methylation and noncoding RNA (e.g., miRNA, lncRNA, circular RNA) and plays a crucial role in several diseases, but most of the studies have involved in vitro experiments [55].
3. Postoperative Cognitive Impairment and Epigenetic Mechanisms
3.1. DNA Methylation
DNA methylation is facilitated by DNA methyltransferases (DNMT1, DNMT3a, DNMT3b) and generally leads to the repression of gene expression through the addition of methyl groups to CpG sites of the gene promoters, which blocks the binding of transcription factors. DNA methylation is one of the most well-described epigenetic modifications, and regulates numerous diverse cellular processes including the silencing of transposable elements, X-chromosome inactivation and tissue-specific gene expression [82].
In the first part of this section, we summarize studies including human subjects. It was demonstrated recently that neurosurgery significantly altered DNA methylation levels of 24 CpG sites of the TNF, IL1B and IL6 gene (Figure 1). In addition, it was shown that the inflammatory methylation index, which was based on the postoperative DNA methylation levels at five selected CpG sites of the genes mentioned above, can be, with moderate accuracy with an area under the curve value of 0.84, a putative detection tool for delirium [83]. Li et al. showed recently that postoperative global hypomethylation of leukocyte DNA is associated with the development of early POCD in elderly patients undergoing hip surgery [84]. A study evaluated the expression and methylation profile of hOGG1 and XRCC1, two important DNA-repair genes, under isoflurane and propofol anesthesia in 40 patients undergoing elective and minimally invasive surgery. Although an increased expression of these genes, depending on surgery time, was detected after isoflurane anesthesia, this was not accompanied by altered methylation in the genes [56].
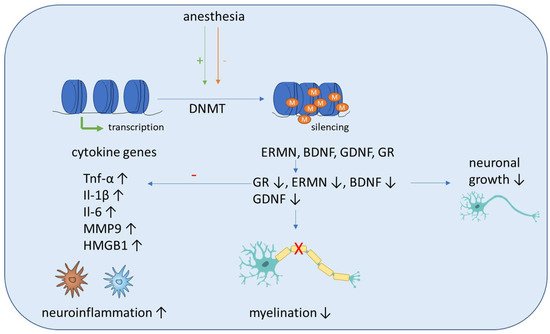
Figure 2. Schematic overview of the molecular mechanisms induced by anesthesia on DNA methylation. Anesthesia can cause both an increase and decrease of DNA-methyltransferase (DNMT) activity. The methylation of inflammatory genes (Tnf-α, IL-1β, Il-6, MMP9, HMGB1) is generally decreased, which leads to increased expression and aggravates neuroinflammation. Anti-inflammation is decreased by decreased expression of the anti-inflammatory transcription factor glucocorticoid receptor (GR). In addition, the methylation of genes associated with neuronal growth (BDNF, GDNF), differentiation and survival of existing neurons (ERMN) is increased, which reduces their expression and causes cognitive impairment due to demyelination and the reduced growth of neurons.
Sadahiro et al. showed that major surgery induces acute changes in DNA methylation in peripheral blood mononuclear cells associated with immune response pathways that might influence the risk of postoperative complications, including cognitive dysfunction and delirium [85]. Shinozaki et al. demonstrated in a collective of neurosurgical patients that a decrease in the DNA methylation of cytokines gene CpGs in glia cells and blood cells can be seen with aging. As this can affect their expression, additional research is needed to fully elucidate the role of DNA methylation in aging and how it may influence the pathogenesis of delirium [86]. Zhang et al. conducted a study including young nonhuman primates, mice and children and proposed a pathway that can lead to demyelination in the brains of young individuals after sevoflurane anesthesia. They showed that blood folate levels are reduced in children after anesthesia and surgery, which causes a downregulation of thymidylate synthase gene after sevoflurane anesthesia. The primary target of this disrupted folate metabolism is the ERM-like protein (ERMN), whose gene is epigenetically altered after sevoflurane. The increased methylation of the ERMN promotor causes decreased ERMN expression and leads to demyelination in the brain, which causes cognitive impairment (Figure 2). Therefore, anesthesia leads to disrupted folate metabolism and then defects in myelination in the developmental brain, and ERMN is the important target affected by the anesthesia via epigenetic mechanisms [87].
In the context of neurological disorders, DNA methylation in astrocytes appears to be altered, which can impact on both the neuroprotective and neurodegenerative qualities of astrocytes and affects astrocyte differentiation and inflammatory response [88].
Summarizing the effects seen after surgery mostly includes pathways which are related to immune response or neuronal growth (Figure 2).
In the second part, we focus on animal studies that might help us to get a deeper view into the pathomechanisms of cognitive impairment induced by certain anesthetics.
3.1.1. Sevoflurane Effects in Animal Studies
It was recently demonstrated that sevoflurane exposure leads to increased DNMT expression in pyramidal neurons in rats, which causes cognitive impairment and is accompanied by hypermethylation of Reelin genes and BDNF, and subsequently their downregulation, which finally leads to a reduction of dendritic spines in the pyramidal neurons of the hippocampus [89,90] (Figure 2).
Zhong et al. demonstrated that sevoflurane-induced POCD in elderly mice induces compromised levels of global DNA 5′-hydroxymethylcytosine and ubiquitin-like effects with PHD and ring finger domains 2 in the hippocampus and the amygdaloid nucleus, when compared with non-POCD and control mice. In addition, the 5-hydroxymethylcytosin reduction of the promoters of genes associated with development and neural protection, such as BDNF, glial cell-derived neurotrophic factor, acyl-CoA synthetase short chain family member 2 and glucocorticoid receptor, activated the transcription of these genes and could be found in the hippocampus of POCD mice. Contrarily, increased 5-methylcytosine levels on the promoters of glutamate receptor 2 precursor GluR2 and decreased 5-methylcytosine levels of HMGB1 and MMP9 showed an inverse tendency with transcription of these genes [91]. Zhu et al. showed that reduced glucocorticoid receptor expression due to upregulated methylation levels of the promoter region of glucocorticoid receptor exon 17 enhances neuroinflammation and triggers cognitive dysfunction after sevoflurane anesthesia in adult rats [92] (Figure 2). Chastain-Potts et al. showed that sevoflurane exposure of rat pups results in decreased neuronal 5-methycytosine, indicating reduced DNA methylation [93].
3.1.2. Isoflurane Anesthesia Effects in Animal Studies
Klenke et al. recently showed that DNA methylation in the DNA of mouse primary hippocampal neurons after isoflurane exposure alters the methylation of the genes chemokine (C-X-C motif) ligand 12, chemokine (C-X-C motif) ligand 14, GATA binding protein 3, IL11, IL13, and IL4 receptor alpha belonging to inflammatory pathway [94]. Gregoire et al. showed that global DNA methylation is reduced after surgery and that the oral administration of the methyl donor S-adenosylmethionine can attenuate sensory and cognitive impairment associated with nerve injury after isoflurane anesthesia in mice. These effects might be mediated partly by the systemic administration of the methyl donor S-adenosylmethionine over modulation of DNA methylation in the CNS [95].
3.2. Histone Acetylation
Histone acetylation and deacetylation are epigenetic processes mediated by histone acetyl transferases and deacetylases (HDACs) [96]. It is generally assumed that the methylation of histone tails can result in either the induction or repression of gene transcription, but acetylation of histone tails is solely associated with active gene expression [96]. Mechanistically, histone deacetylation enhances the binding of histones to DNA and the aggregation of chromosomes, resulting in transcription inhibition, and is connected to heterochromatin, while histone acetylation is connected to transcriptional activation and associated with euchromatin. It is well-known that inflammation and anesthetics can also influence histone acetylation [54,97,98]. Interestingly, HDAC inhibitory activity can be found in drugs with known anti-inflammatory and neuroprotective functions, such as valproic acid [99]. Here, we summarize the effects of distinct anesthetics on histone acetylation. As little is known about the impact of propofol and midazolam on histone acetylation, these drugs may be superior compared to isoflurane and sevoflurane, where several effects on histone acetylation are known.
3.2.1. Propofol-Induced Effects
A recent study indicated that propofol might be involved in the regulation of histone acetylation in cancer development. Holownia et al. showed that propofol regulates histone acetylation in rat astroglial cells by protection against tert-butyl hydroperoxide toxicity [72]. Lin et al. proved that propofol application during early gestation could affect the offspring’s learning and memory by inhibiting histone acetylation [100].
3.2.2. Midazolam-Induced Effects
In 2016, it was demonstrated in rat hippocampus after exposure to a sedative dose of midazolam followed by combined nitrous oxide and isoflurane, that anesthesia causes epigenetic modulations manifested as histone-3 hypoacetylation, with a 25% decrease in histone acetyltransferase activity. This was accompanied by a downregulation of the transcription of BDNF and cellular Finkel-Biskis-Jinkins murine sarcoma virus osteosarcoma oncogene. Hence, the authors postulated that long-term impairments of neuronal development and synaptic communication could be caused by anesthesia-induced epigenetic phenomena [98].
3.2.3. Isoflurane-Induced Effects
The incubation of THP-1 monocytes with isoflurane results in increased HDAC1 and HDAC2 expression and downregulates cytokine expression. These results were shown in primary human peripheral blood monocytes, where HDAC1 and HDAC2 gene silencing caused increased cytokine production and NF-κB nuclear translocation induced by isoflurane pre-exposure and lipopolysaccharide stimulation. These findings showed that anti-inflammatory effects of isoflurane in human monocytes involve the regulation of HDAC1 and HDAC2 [101]. These results are in contrast to those of studies with rats, which showed that isoflurane exposure increased cognitive impairment significantly. Here an upregulation of HDAC 2, reduction of histone acetylation of AcH4K12 and AcH3K9, and increase inflammation (Il-1β, Il-6) (Figure 3) and apoptosis in the hippocampus were shown. Additional impairments of BDNF-tyrosine kinase receptor B (TrkB) signaling and the downstream signaling pathway including phospho-calmodulin-dependent protein kinase and phospho-cAMP response element-binding protein. These findings indicate that isoflurane-induced cognitive dysfunction is associated with the downregulation of the BDNF-TrkB signaling pathway due to declines in chromatin histone acetylation [97]. Hence both above mentioned studies have in common that HDAC2 expression is increased after isoflurane exposure, even if its impact on cytokine expression is contrary. In addition, mice that are neonatally exposed to isoflurane show significant memory impairment and exhibit dysregulated hippocampal H4K12 acetylation and decreased c-Fos expression, which can be attenuated by the HDACi trichostatin A [102]. Jin et al. showed that enhancing the expression of antiapoptotic proteins (Bcl-2, Bcl-xL, xIAP, c-IAP-1, c-IAP-2 and survivin) and improving the acetylation of H3K9 and H4K12 improves the cognitive function of rats after isoflurane anesthesia via the downregulation of the expression of the HDACs HDAC2 and HDAC-3 [103]. Chen et al. demonstrated that isoflurane exposure in aged rats leads to impaired spatial learning and memory, accompanied by dysregulated H3K9 and H4K12 acetylation, which is, in turn, accompanied by reduced BDNF expression and the suppression of the BDNF downstream signaling pathway. Restoring histone acetylation and BDNF signaling suppresses proinflammatory cytokines and the NFκB signaling pathway and attenuates isoflurane-induced cognitive dysfunction [64]. It was demonstrated recently that isoflurane anesthesia increases the expression of HDAC3 protein and decreases levels of dendritic spine density and synaptic plasticity-related proteins in the dorsal hippocampus of aged mice [104].
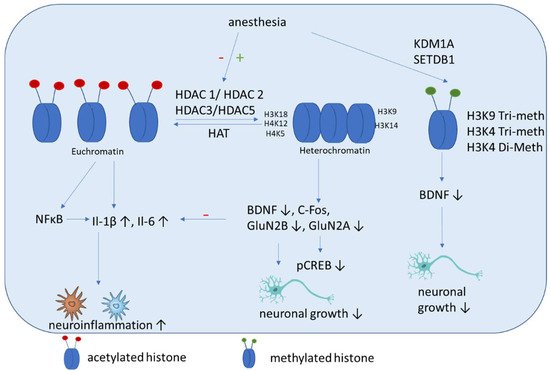
Figure 3. Schematic overview of the molecular mechanisms induced by anesthesia on histone modifications. Anesthesia can cause both a decrease of histone acetylation and an increase of HDAC activity. Histone deacetylation mainly causes an increase of inflammatory cytokines and a decrease of factors associated with neuronal growth, differentiation and survival of existing neurons. In addition, methylation (SETDB1) and demethylation (KDM1A) are induced by anesthesia, which can cause a decrease of BDNF.
Zhong et al. showed that neonatal isoflurane exposure-induced memory impairment is associated with the dysregulation of H4K12 acetylation, which may lead to less hippocampal activation and cognitive impairment in mice [105].
3.2.4. Sevoflurane-Induced Effects
Sevoflurane impairs synaptic plasticity and cognitive function in aged mice [106]. It was demonstrated recently that sevoflurane inhibits histone acetylation at H3K18, H3K14, H4K5 and H4K12 (Figure 3). This can induce decreased binding of H3K18 to the promoter of GluN2B and H3K14 to the promoter of the GluN2A gene. In addition, sevoflurane increases ANP32A expression by activating C/EBPβ [107]. All these effects are associated with cognitive impairment.
The cognitive dysfunction after sevoflurane anesthesia can be alleviated by HDACi trichostatin A treatment via inhibition of the overactivation of astrocyte intracellular NF-κB signaling and the release of cytokines [92]. Histone deacetylases modulate cytokine synthesis and release. In line with this, trichostatin A diminishes lipopolysaccharide-induced inflammatory responses in the mouse brain and modulates t cytokine-associated changes in cognitive function, which may be specifically related to reducing HDAC2 and HDAC5 expression [108]. Zhang et al. showed that inhibition of HDAC significantly reverses the sevoflurane-induced decrease in Ac-H3, BDNF, tropomyosin-related kinase B and p-cAMP response element-binding expressions, and reduces the level of apoptosis-related protein-cleaved caspase-3. They conclude that normalizing the hippocampal histone acetylation state might resolve cognitive and synaptic plasticity impairments induced by sevoflurane exposure [109].
5.3. Histone Methylation
The role of the methylation of histones facilitated by histone methylases is not yet fully understood. It results in either increased or reduced gene expression, depending on the specific amino acid methylated on the histone tails. It is assumed that H3K4 tri-methylation is associated with transcription increase, whereas H3K9 tri-methylation leads to reduced transcription [110].
Histone methylation has not been studied well in the context of cognitive impairment. Wu et al. showed that histone H3K9 trimethylation downregulates the expression of BDNF in the dorsal hippocampus and impairs memory formation during anesthesia with laparotomy under isoflurane inhalation and surgery in mice [111]. Our group showed recently that propofol decreased H3K4 tri-methylation, and that cholinergic genes bind to this histone [112]. In addition, we demonstrated that midazolam increases the expression of lysine-specific demethylase (KDM1A), which is accompanied by a decreased di-methylation of H3K4 [113]. In addition, a recent network biology approach identified that histone-lysine N-methyltransferase SETDB1, among other genes, is upregulated in patients with POCD [114].
Other histone modifications exist in addition to methylation and acetylation, and include phosphorylation, ubiquitylation and sumoylation. However, not much is known about these modifications in the context of cognitive function, but existing studies indicate that astrocyte-specific histone phosphorylation, ubiquitylation and sumoylation could be important factors to examine in neurodegenerative diseases [115].
This entry is adapted from the peer-reviewed paper 10.3390/cells11192954
This entry is offline, you can click here to edit this entry!