Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Cisplatin (CDDP) is the drug of choice against different types of cancer. However, tumor cells can acquire resistance to the damage caused by cisplatin, generating genetic and epigenetic changes that lead to the generation of resistance and the activation of intrinsic resistance mechanisms in cancer cells.
- cancer
- cisplatin
- drug resistance
- epigenetics
1. Introduction
Cancer is an important cause of morbidity and mortality worldwide, in every region, and irrespective of the level of human development. It has been reported that in 2020, about 9.9 million cancer deaths occurred worldwide. Studies indicate that new cancer cases will increase from 19.3 to 28.4 million by 2040 [1][2]. Cancer pathology has genetic, inflammatory, and metabolic components, which are presented by the sequential accumulation of mutations in the genome and lead to the acquisition of the tumor phenotype characterized by metabolic alterations, high proliferation rates, resistance to apoptosis, and growth factor independence, among others [3]. Cancer originates from gathering molecular alterations of genetic and/or epigenetic origin. These can be initiated by the accumulation of genetic DNA damage, affecting the DNA sequence (such as mutations and chromosomal rearrangements) or modifications in DNA, histones, and non-coding RNA that do not lead to a change in the original sequence (epigenetic modifications) [4].
Cisplatin (cis-diamminedichloroplatinum (II), CDDP) is currently the treatment of choice for many types of cancer [5][6][7][8][9][10][11]. Cisplatin exerts anticancer activity via multiple mechanisms. Its most acceptable mechanism involves the formation of DNA–platinum adducts by interacting with purine bases, activating several signal transduction pathways, and silencing or activating several genes which finally leads to apoptosis. However, side effects and drug resistance are the two inherent challenges of cisplatin that limit its application and effectiveness. The reduction of drug accumulation inside cancer cells, inactivation of drugs by reacting with glutathione and metallothioneins, and faster repairing of DNA lesions are responsible for cisplatin resistance [12].
2. Cisplatin: Mechanism of Action
Cisplatin is a neutral coordination complex with a central platinum (II) atom bonded to two chloride atoms and two ammonia molecules in the cis position. The coordinated covalent bonds of platinum with nitrogen are virtually irreversible, but their bonds with chloride ligands, in aqueous media and under certain pH and temperature conditions, are highly labile [13].
Cisplatin’s mechanism of action is initiated by the activation of the complex in the intracellular medium by the hydrolysis of chloride molecules. The cisplatin molecule hydrolyzes in the cytoplasm, and acts as a potent electrophilic agent, reacting with nucleic acids and sulfhydryl groups of proteins [14][15]. However, the therapeutic target of this drug is genomic and mitochondrial DNA. The covalent binding of CDDP to DNA via platinum atoms, by intercalating between base pairs (mainly purines), generates so-called cisplatin–DNA adducts. Platinum binds mainly through nitrogen at position 7 of the imidazole ring of the guanine and adenine of the corresponding DNA nucleotides (2′-deoxyadenosine 5′-monophosphate, dAMP; and 2′-deoxyguanosine 5′-monophosphate, dGMP) since these are the atoms with the highest electron density, and are most accessible to electrophilic attack by cisplatin. Moreover, binding is particularly favored with guanines located in the major groove of the DNA double helix [14][16][17]. As a consequence of the formation of these DNA adducts, the DNA replication mechanisms will be inhibited and therefore effect its transcription processes [13]. In response to this cellular damage, signaling pathways will be activated that will lead in the first instance to cell cycle arrest through the action of the tumor suppressor protein p53 in an attempt to repair the damaged DNA [18][19]. Subsequently, cell death by apoptosis occurs mediated by proteins such as Bcl-2 if the DNA damage is not repaired [14][18].
3. Resistance to Cisplatin Treatment
3.1. Mechanisms of Cisplatin Resistance
The development of chemotherapeutic resistance is a problem of great importance despite great advances in understanding the molecular mechanisms of cancer [20][21]. It has been observed that 50% of patients treated with cisplatin either go on to develop intrinsic resistance or acquire multidrug resistance rapidly [13][22][23]. In both cases, the mechanisms of resistance are based on a reduction in the accumulation of cytotoxic compounds in the cytosol of cancer cells, together with the activation of DNA repair mechanisms that protect cancer cells from potentially lethal stresses caused by chemo drugs [24].
A cell population is considered to be resistant when it increases its baseline tolerance, managing to proliferate in a medium with twice, or more than twice, the drug concentration tolerated by the parental line, for which mechanisms are activated that allow it to avoid drug-induced cell death, which is related to morphological variations described as an increase in cell size, increase in the nucleus–cytoplasmic ratio, irregularities in the cell membrane borders, or an increase in cytoplasmic granules [25][26][27].
Resistance to CDDP and other chemo drugs are directly related to the stage of tumor progression because cancer cells acquire additional genetic and epigenetic alterations that confer growth advantages, such as proliferation, and consequently, the expected cytotoxic or cytostatic effect does not occur [28]. Both mutations and changes in gene expression and post-translational modifications of proteins are some of the alterations that have been associated with the acquisition of resistance to these drugs [27][29][30].
3.1.1. Pre-Target Resistance
Pre-target resistance is related to the reduction of CDDP entry into the cell or to a more significant expulsion of CDDP into the extracellular space [16][31]. CDDP is a very polar molecule and enters cells relatively slowly compared to other molecules used for cancer treatment. CDDP entry into the cell is influenced by the concentrations of sodium and potassium ions, pH, the presence of reducing agents, and the action of transporters and channels, which are coupled to the passive diffusion mechanism [16]. Among the proposed transporters, the organic cation transporters (OCT) and the copper transporter protein CTR1 (copper transport protein 1) stand out. It was observed that cisplatin causes a decrease in the expression of these transporter proteins, decreasing the concentration of the drug inside the cells as a mechanism of resistance [32][33][34][35][36]. On the other hand, some studies suggest that the transporter proteins ATP7A and ATP7B and the multidrug resistance-associated protein MRP2 may also be involved in CDDP resistance by increasing the flux of CDDP out of the cell [35][37][38][39]. Another “pre-target” mechanism refers to the intracellular inactivation of cisplatin by the formation of complexes with compounds present in the cell cytosol, mainly those containing thiol groups such as reduced glutathione (GSH) or metallothioneins. This process occurs in the cytoplasm where cisplatin is a potent electrophilic agent that acts with these nucleophilic groups and thus decreases drug interactions with DNA [40][41].
3.1.2. On-Target Resistance
On-target resistance involves processes related to molecular damage caused by cisplatin to DNA [16][31]. Once CDDP is bound to DNA, the cell can survive by activating DNA repair mechanisms or by tolerance to genetic damage. Nucleotide excision repair is the first pathway that begins to repair DNA in the face of cisplatin resistance. This repair pathway is responsible for removing the bonds formed between platinum and DNA. Once CDDP binds to DNA, the cell can survive by activating DNA repair mechanisms or by tolerance to genetic damage. Within the DNA repair pathways, nucleotide excision repair appears to play a key role in eliminating cisplatin damage. This repair pathway is responsible for eliminating the bonds formed between platinum and DNA through the action of ERCC1 (excision repair cross-complementing 1) and XPF (Xeroderma pigmentosum complementation group F) proteins. These proteins form a heterodimer and act by cutting the 5′ end of the area of the strand where the platinum has bound to the DNA to allow subsequent elimination of the adduct. A relationship between increased expression levels of ERCC1 endonuclease and CDDP resistance has been described in different cell lines and patient samples [42][43][44][45][46]. In addition, increased tolerance to cisplatin-induced damage may be related to a loss of function of the mispaired base repair (MMR) pathway. During MMR, different proteins recognize intracatenary adducts, including MSH2 and MLH1, which, together with other MMR proteins, detect damage and transmit proapoptotic signals. MSH2 and MLH1 genes have been mutated or downregulated due to CDDP resistance, resulting in the inhibition of apoptosis [16]. On the other hand, cisplatin induces intercatenary adducts that are usually repaired by the homologous recombination mechanism (HRR). In breast and ovarian cancer, the BRCA1 and BRCA2 genes, which code for proteins of the HRR system, have been found to be mutated [47]. In particular, cancers deficient in the HRR system have a different phenotype and are often more sensitive to cisplatin than their counterparts in which the HRR mechanism functions usually [48]. Finally, it should be mentioned that damage tolerance is related to the replicative by-pass of CDDP-induced injury that certain classes of polymerases, such as β, ƞ, and ζ, can perform. This results in DNA synthesis not being blocked and, consequently, apoptotic pathways are not activated [16][49][50].
3.1.3. Post-Target Resistance
Post-target resistance includes mechanisms that affect signaling pathways leading to cell death triggered by adducts [16][31]. Among these mechanisms is the inactivation of the TP53 gene, which produces a loss of apoptotic activity and the appearance of resistance in 50% of human cancers [51]. TP53 encodes for the p53 protein, which induces apoptosis by activating the signaling cascade to effector molecules such as Bax (BCL2-associated X protein). Similarly, the inactivation of caspases such as caspases 3, 8, and 9, of great importance in apoptosis, has been associated with resistance to cisplatin in different types of cancers such as head and neck, ovarian, breast, and others [52][53][54][55][56]. Cisplatin resistance is also caused by CYLD (CYLD lysine 63 deubiquitinase) downregulation, which triggers the reduction of intracellular CDDP accumulation and the suppression of cell death via NF-κB hyperactivation [57]. TNF-α also contributes to NF-κΒ activation in head and neck cancer cells [58]. Even more, the inhibition of both NF-κΒ and MAPK/HO-1 signaling pathways also reduce oxidative stress and CDDP-induced resistance in non-small cell lung cancer [59] (Figure 1).
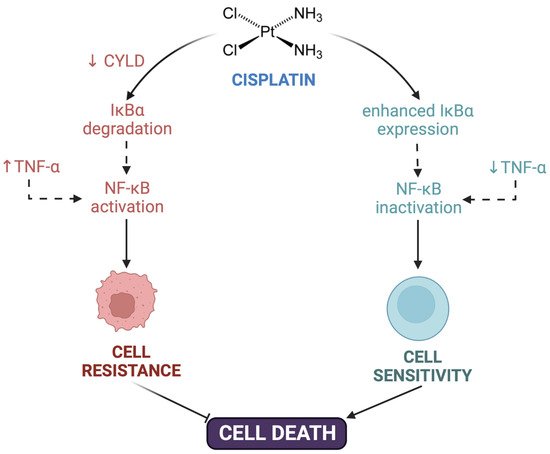
Figure 1. Contribution of NF-κΒ to cisplatin resistance. Cisplatin resistance is caused by downregulation of CYLD lysine 63 deubiquitinase (CYLD), triggering the suppression of cell death via NF-κB hyperactivation. TNF-α also contributed to NF-κΒ activation and cell resistance. Created with Biorender.com.
3.1.4. Off-Target Resistance
Off-target resistance is related to alterations in signaling pathways that are not directly related to cisplatin but interfere with cisplatin-induced proapoptotic events [16][31]. This type of mechanism includes the overexpression of the proto-oncogene ERBB2 that encodes for the HER2 (human epidermal growth factor receptor) protein, and the gene encoding the DYRK1B (dual specificity tyrosine phosphorylation regulated kinase 1B) kinase. The former is key to activating numerous signaling pathways that regulate functions such as cell differentiation, growth, and survival [60]. The second facilitates cell survival by increasing the activity of antioxidant enzymes such as ferroxidase and superoxide dismutase, which constitute the defense of cells against oxidative stress [61]. There are also several mechanisms associated with the organism’s response to stressful situations or poorly characterized ones related to resistance to cisplatin, including autophagy (a cellular process responsible for the degradation and recycling of damaged cellular components) [62][63]. In this sense, different studies postulate that the inhibition of autophagy can restore cell sensitivity to cisplatin, at least in vitro [63].
This entry is adapted from the peer-reviewed paper 10.3390/biom12101365
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249.
- WHO. World Health Statistics 2021: Monitoring Health for the SDGs, Sustainable Development Goals; World Health Organization: Geneva, Switzerland, 2021.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380.
- Vermorken, J.B.; Remenar, E.; Van Herpen, C.; Gorlia, T.; Mesia, R.; Degardin, M.; Stewart, J.S.; Jelic, S.; Betka, J.; Preiss, J.H. Cisplatin, fluorouracil, and docetaxel in unresectable head and neck cancer. N. Engl. J. Med. 2007, 357, 1695–1704.
- Motzer, R.J.; Sheinfeld, J.; Mazumdar, M.; Bajorin, D.F.; Bosl, G.J.; Herr, H.; Lyn, P.; Vlamis, V. Etoposide and cisplatin adjuvant therapy for patients with pathologic stage II germ cell tumors. J. Clin. Oncol. 1995, 13, 2700–2704.
- Li, D.; Zhang, Y.; Xie, Y.; Xiang, J.; Zhu, Y.; Yang, J. Enhanced tumor suppression by adenoviral PTEN gene therapy combined with cisplatin chemotherapy in small-cell lung cancer. Cancer Gene Ther. 2013, 20, 251–259.
- Magali, L.; Pascal, F.; Serge, A.; Mathieu, B.; Ayoube, Z.; Claire, T.; Christiane, M. Better survival in impaired renal function patients with metastatic non-small cell lung cancer treated by cisplatin-pemetrexed. Eur. J. Clin. Pharmacol. 2020, 76, 1573–1580.
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43.
- Moore, K.N.; Herzog, T.J.; Lewin, S.; Giuntoli, R.L.; Armstrong, D.K.; Rocconi, R.P.; Spannuth, W.A.; Gold, M.A. A comparison of cisplatin/paclitaxel and carboplatin/paclitaxel in stage IVB, recurrent or persistent cervical cancer. Gynecol. Oncol. 2007, 105, 299–303.
- Coppin, C.; Gospodarowicz, M.K.; James, K.; Tannock, I.F.; Zee, B.; Carson, J.; Pater, J.; Sullivan, L.D. Improved local control of invasive bladder cancer by concurrent cisplatin and preoperative or definitive radiation. The National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 1996, 14, 2901–2907.
- Yu, Z.; Cao, W.; Ren, Y.; Zhang, Q.; Liu, J. ATPase copper transporter A, negatively regulated by miR-148a-3p, contributes to cisplatin resistance in breast cancer cells. Clin. Transl. Med. 2020, 10, 57–73.
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378.
- Basu, A.; Krishnamurthy, S. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010, 201367.
- Florea, A.-M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371.
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883.
- Shen, D.-W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721.
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279.
- Fuertes, M.; Castilla, J.; Alonso, C.; Prez, J. Cisplatin biochemical mechanism of action: From cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr. Med. Chem. 2003, 10, 257–266.
- Kartal-Yandim, M.; Adan-Gokbulut, A.; Baran, Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit. Rev. Biotechnol. 2016, 36, 716–726.
- Steding, C.E. Creating chemotherapeutic-resistant breast cancer cell lines: Advances and future perspectives. Future Oncol. 2016, 12, 1517–1527.
- Pogribny, I.P.; Filkowski, J.N.; Tryndyak, V.P.; Golubov, A.; Shpyleva, S.I.; Kovalchuk, O. Alterations of microRNAs and their targets are associated with acquired resistance of MCF-7 breast cancer cells to cisplatin. Int. J. Cancer 2010, 127, 1785–1794.
- Velasco, G.; Sánchez, C.; Guzmán, M. Anticancer mechanisms of cannabinoids. Curr. Oncol. 2016, 23, 23–32.
- Aye, Y.; Li, M.; Long, M.; Weiss, R. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2015, 34, 2011–2021.
- McDermott, M.; Eustace, A.; Busschots, S.; Breen, L.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro development of chemotherapy and targeted therapy drug-resistant cancer cell lines: A practical guide with case studies. Front. Oncol. 2014, 4, 40.
- Lukyanova, N.Y.; Rusetskya, N.; Tregubova, N.; Chekhun, V. Molecular profile and cell cycle in MCF-7 cells resistant to cisplatin and doxorubicin. Exp. Oncol. 2009, 31, 87–91.
- Puspita, N.A.; Bedford, A. Morphological Changes of Cisplatin-resistant Human Breast Cancer MCF-7 Cell Line. Int. J. Integr. Health Sci. 2017, 5, 8–14.
- Hinojosa-García, L.M.; Dueñas-González, A. Papel de la quimioterapia en el tratamiento del carcinoma cervicouterino. Rev. Inst. Nal. Cancerol. (Mex) 2000, 46, 47–57.
- Aleksakhina, S.N.; Kashyap, A.; Imyanitov, E.N. Mechanisms of acquired tumor drug resistance. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2019, 1872, 188310.
- Lund, R.J.; Huhtinen, K.; Salmi, J.; Rantala, J.; Nguyen, E.V.; Moulder, R.; Goodlett, D.R.; Lahesmaa, R.; Carpén, O. DNA methylation and transcriptome changes associated with cisplatin resistance in ovarian cancer. Sci. Rep. 2017, 7, 1469.
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257.
- Müller, J.; Lips, K.S.; Metzner, L.; Neubert, R.H.; Koepsell, H.; Brandsch, M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT). Biochem. Pharmacol. 2005, 70, 1851–1860.
- Ciarimboli, G. Organic Cation Transporters 2 as Mediators of Cisplatin Nephrotoxicity. In Platinum and Other Heavy Metal Compounds in Cancer Chemotherapy; Bonetti, A., Leone, R., Muggia, F.M., Howell, S.B., Eds.; Cancer Drug Discovery and Development. Humana Press: Totowa, NJ, USA, 2009; pp. 353–358.
- Safaei, R. Role of copper transporters in the uptake and efflux of platinum containing drugs. Cancer Lett. 2006, 234, 34–39.
- Kuo, M.T.; Fu, S.; Savaraj, N.; Chen, H.H. Role of the human high-affinity copper transporter in copper homeostasis regulation and cisplatin sensitivity in cancer chemotherapy. Cancer Res. 2012, 72, 4616–4621.
- Köberle, B.; Brenner, W.; Albers, A.; Usanova, S.; Thüroff, J.W.; Kaina, B. ERCC1 and XPF expression in human testicular germ cell tumors. Oncol. Rep. 2010, 23, 223–227.
- Liang, Z.D.; Long, Y.; Tsai, W.-B.; Fu, S.; Kurzrock, R.; Gagea-Iurascu, M.; Zhang, F.; Chen, H.H.; Hennessy, B.T.; Mills, G.B. Mechanistic basis for overcoming platinum resistance using copper chelating agents. Mol. Cancer Ther. 2012, 11, 2483–2494.
- Korita, P.V.; Wakai, T.; Shirai, Y.; Matsuda, Y.; Sakata, J.; Takamura, M.; Yano, M.; Sanpei, A.; Aoyagi, Y.; Hatakeyama, K. Multidrug resistance-associated protein 2 determines the efficacy of cisplatin in patients with hepatocellular carcinoma. Oncol. Rep. 2010, 23, 965–972.
- Yamasaki, M.; Makino, T.; Masuzawa, T.; Kurokawa, Y.; Miyata, H.; Takiguchi, S.; Nakajima, K.; Fujiwara, Y.; Matsuura, N.; Mori, M. Role of multidrug resistance protein 2 (MRP2) in chemoresistance and clinical outcome in oesophageal squamous cell carcinoma. Br. J. Cancer 2011, 104, 707–713.
- Chen, H.H.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met.-Based Drugs 2010, 2010, 430939.
- Knipp, M.; Karotki, A.V.; Chesnov, S.; Natile, G.; Sadler, P.J.; Brabec, V.; Vašák, M. Reaction of Zn7Metallothionein with cis-and trans- anticancer complexes: Trans-PtII complexes retain their N-donor ligands. J. Med. Chem. 2007, 50, 4075–4086.
- Martin, L.P.; Hamilton, T.C.; Schilder, R.J. Platinum resistance: The role of DNA repair pathways. Clin. Cancer Res. 2008, 14, 1291–1295.
- Saldivar, J.S.; Wu, X.; Follen, M.; Gershenson, D. Nucleotide excision repair pathway review I: Implications in ovarian cancer and platinum sensitivity. Gynecol. Oncol. 2007, 107, S56–S71.
- Chang, I.-Y.; Kim, M.-H.; Kim, H.B.; Kim, S.-H.; Kim, H.-Y.; You, H.J. Small interfering RNA-induced suppression of ERCC1 enhances sensitivity of human cancer cells to cisplatin. Biochem. Biophys. Res. Commun. 2005, 327, 225–233.
- Usanova, S.; Piée-Staffa, A.; Sied, U.; Thomale, J.; Schneider, A.; Kaina, B.; Köberle, B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol. Cancer 2010, 9, 248.
- Hirakawa, M.; Sato, Y.; Ohnuma, H.; Takayama, T.; Sagawa, T.; Nobuoka, T.; Harada, K.; Miyamoto, H.; Sato, Y.; Takahashi, Y. A phase II study of neoadjuvant combination chemotherapy with docetaxel, cisplatin, and S-1 for locally advanced resectable gastric cancer: Nucleotide excision repair (NER) as potential chemoresistance marker. Cancer Chemother. Pharmacol. 2013, 71, 789–797.
- Narod, S.A.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer 2004, 4, 665–676.
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921.
- Bassett, E.; Vaisman, A.; Tropea, K.A.; McCall, C.M.; Masutani, C.; Hanaoka, F.; Chaney, S.G. Frameshifts and deletions during in vitro translesion synthesis past Pt–DNA adducts by DNA polymerases β and η. DNA Repair 2002, 1, 1003–1016.
- Albertella, M.R.; Green, C.M.; Lehmann, A.R.; O’Connor, M.J. A role for polymerase η in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005, 65, 9799–9806.
- Martinez-Rivera, M.; Siddik, Z.H. Resistance and gain-of-resistance phenotypes in cancers harboring wild-type p53. Biochem. Pharmacol. 2012, 83, 1049–1062.
- Ding, Z.; Yang, X.; Pater, A.; Tang, S.-C. Resistance to apoptosis is correlated with the reduced caspase-3 activation and enhanced expression of antiapoptotic proteins in human cervical multidrug-resistant cells. Biochem. Biophys. Res. Commun. 2000, 270, 415–420.
- Duiker, E.W.; Meijer, A.; van der Bilt, A.R.; Meersma, G.J.; Kooi, N.; van der Zee, A.G.; De Vries, E.; de Jong, S. Drug-induced caspase 8 upregulation sensitises cisplatin-resistant ovarian carcinoma cells to rhTRAIL-induced apoptosis. Br. J. Cancer 2011, 104, 1278–1287.
- Kim, P.K.; Mahidhara, R.; Seol, D.-W. The role of caspase-8 in resistance to cancer chemotherapy. Drug Resist. Updates 2001, 4, 293–296.
- Kuwahara, D.; Tsutsumi, K.; Oyake, D.; Ohta, T.; Nishikawa, H.; Koizuka, I. Inhibition of caspase-9 activity and Apaf-1 expression in cisplatin-resistant head and neck squamous cell carcinoma cells. Auris Nasus Larynx 2003, 30, 85–88.
- Nikounezhad, N.; Nakhjavani, M.; Shirazi, F.H. Generation of cisplatin-resistant ovarian cancer cell lines. Iran. J. Pharm. Sci. 2016, 12, 11–20.
- Suenaga, N.; Kuramitsu, M.; Komure, K.; Kanemaru, A.; Takano, K.; Ozeki, K.; Nishimura, Y.; Yoshida, R.; Nakayama, H.; Shinriki, S.; et al. Loss of Tumor Suppressor CYLD Expression Triggers Cisplatin Resistance in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 5194.
- Kim, S.B.; Kim, J.S.; Lee, J.H.; Yoon, W.J.; Lee, D.S.; Ko, M.S.; Kwon, B.S.; Choi, D.H.; Cho, H.R.; Lee, B.J.; et al. NF-kappaB activation is required for cisplatin-induced apoptosis in head and neck squamous carcinoma cells. FEBS Lett. 2006, 580, 311–318.
- Wang, L.H.; Li, Y.; Yang, S.N.; Wang, F.Y.; Hou, Y.; Cui, W.; Chen, K.; Cao, Q.; Wang, S.; Zhang, T.Y.; et al. Gambogic acid synergistically potentiates cisplatin-induced apoptosis in non-small-cell lung cancer through suppressing NF-kappaB and MAPK/HO-1 signalling. Br. J. Cancer 2014, 110, 341–352.
- Deng, X.; Ewton, D.Z.; Friedman, E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res. 2009, 69, 3317–3324.
- Fijołek, J.; Wiatr, E.; Rowińska-Zakrzewska, E.; Giedronowicz, D.; Langfort, R.; Chabowski, M.; Orłowski, T.; Roszkowski, K. p53 and HER2/neu expression in relation to chemotherapy response in patients with non-small cell lung cancer. Int. J. Biol. Markers 2006, 21, 81–87.
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838.
- Ren, J.-H.; He, W.-S.; Nong, L.; Zhu, Q.-Y.; Hu, K.; Zhang, R.-G.; Huang, L.-L.; Zhu, F.; Wu, G. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother. Radiopharm. 2010, 25, 75–80.
This entry is offline, you can click here to edit this entry!