Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Enhancers are distal cis-acting elements that are commonly recognized to regulate gene expression via cooperation with promoters. Along with regulating gene expression, enhancers can be transcribed and generate a class of non-coding RNAs called enhancer RNAs (eRNAs).
- enhancer RNA (eRNA)
- cancer
- prognosis
- diagnosis
1. Introduction
Enhancers are distal cis-acting elements that are known to regulate gene expression via spatial chromatin loops formation with target promoters [1][2]. They are short (50–1500 bp) regulatory elements of accessible DNA that assist in regulating the cell transcriptional machinery through increasing the transcription of target genes. Structurally, enhancers are open/accessible chromatin with low levels of DNA methylation, which are bound by RNA polymerase II (RNApol II), transcription factors (TFs), and cofactors, particularly transcription initiation factors, such as TBP, TFII, and P300/CBP. Enhancers are flanked by histones with permissive chromatin markers of histone H3 lysine 27 acetylation (H3K27ac) and histone H3 lysine 4 methylation (H3K4me) [3][4][5][6][7]. While promoters are cis-acting elements that recruit transcription in a position- and direction-dependent manner, enhancers perform freely of their position and orientation regarding their target gene; consequently, these elements can establish physical communication to interact distant promoters. Rather than contributing to gene expression, enhancers can be dynamically transcribed, forming a class of non-coding RNAs known as enhancer RNAs (eRNAs). It was initially anticipated that the product of enhancer transcription is the noisy outcome of the transcription procedure. Nevertheless, later studies suggest various roles for eRNA as a universal cellular mechanism involved in directing cell characteristics and function. In this research, researchers demonstrate recent understanding of eRNA structure along with function.
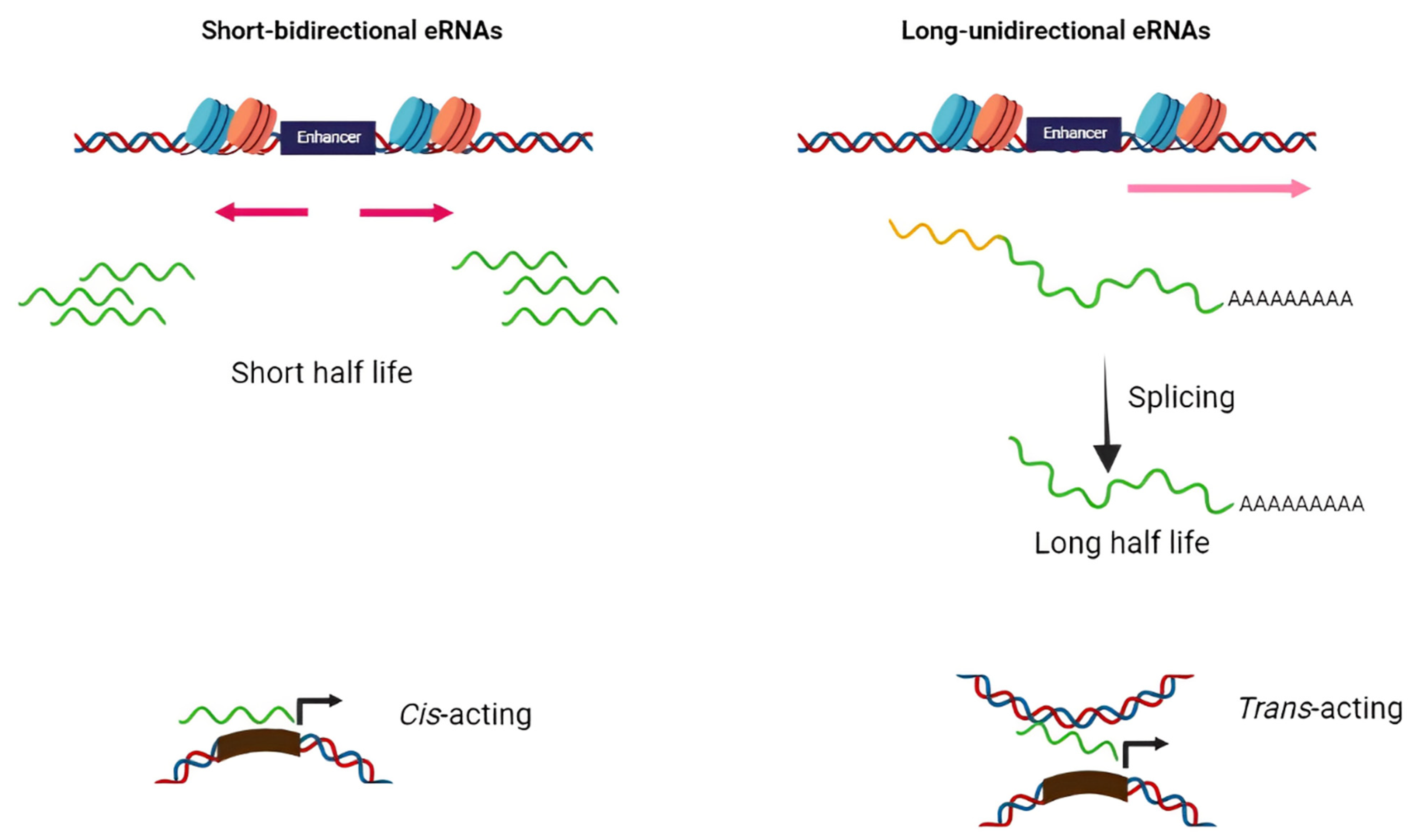
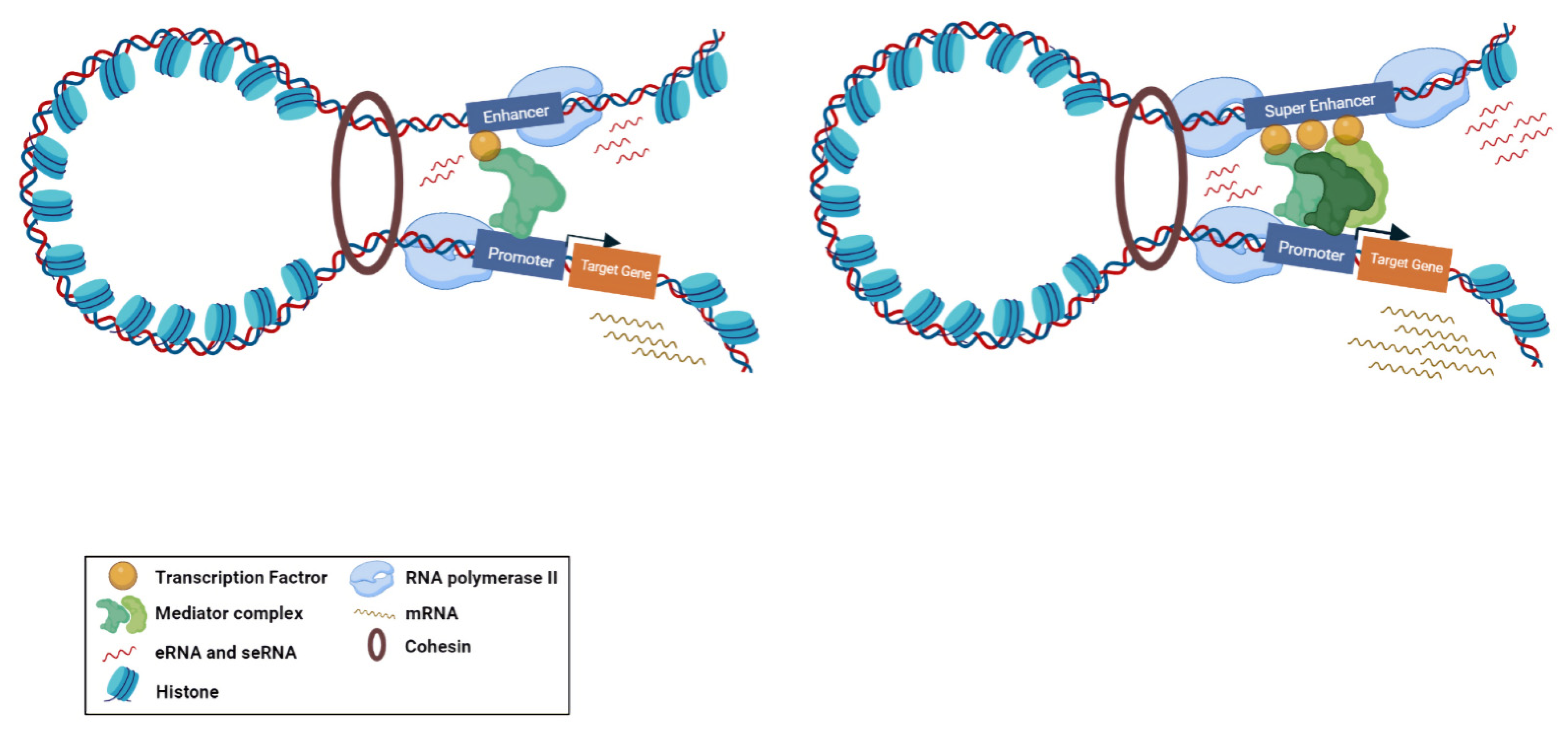
2. Biogenesis and Function of eRNA
Based on structure and transcription patterns, eRNAs (approximately from 0.1–9 Kb) [8] can be classified in two groups of short bidirectional, non-spliced, non-polyadenylated RNAs and long unidirectional, spliced, stable polyadenylated transcripts (Figure 1). Since eRNAs are mainly non-polyadenylated and unstable, they are predominantly localized in the nucleus and chromatin-enriched fractions [9][10][11][12]. Transcription of eRNAs generally occurs prior to mRNA expression from the target gene [13][14][15][16]. The tissue-specific transcription of enhancers has been shown in various diseases such as cancers. Enhancers typically contain specific DNA elements that are recognized by tissue-specific TFs. These factors often cooperate in their binding to enhancers and frequently synergize to achieve the optimal activation of target genes [17]. Under extracellular stimuli and the activation of specific signaling pathways, TFs are recruited into the enhancer region, bind to particular DNA sequences, and stimulate the remodeling of nucleosome and histone modifications (regions enriched by H4K8ac, H3K27ac, and H3K4me are hallmarks of active enhancers) [18][19][20]. H3K27 and H4K8 are acetylated through CBP histone acetyltransferases, and p300 and chromatin is further opened in the enhancer region and, thus, RNApol II and BRD4 cofactor are recruited to the enhancer [20]. Integrator, a large complex associated with the carboxyl-terminal domain (CTD) of RNApol II, has an important role in transcriptional termination at the enhancers. The depletion of the integrator leads to the reduction in processed eRNAs and accumulation of primary eRNA transcripts [21].
Figure 1. Schematic diagram of two distinct classes of eRNAs. The majority of eRNAs are short, bidirectional, non-polyadenylated, and unstable while others are long unidirectional, polyadenylated, and more stable. The former has cis-acting action while the latter perform as trans-acting elements.
Super-enhancers (SE) are described as a cluster of enhancers that have dense assemblies of RNApol II, TFs, and typical enhancer histone modifications (H4K8ac, H3K27ac, and H3K4me) that leads to a greater amount of super-enhancer RNA (seRNA) production (Figure 2) [22]. The difference between conventional enhancers and SEs is clearly displayed in the nature of the dependence of the transcription activity ensured by the regulatory element and the number of TFs and cofactors associated with it [23]. The transcription activity at an SE is typically higher than at a distinct enhancer. SEs have high potential to activate the transcription of their target genes and play significant roles in tissue-specific biological processes [24]. Most SE produce unidirectional polyadenylated seRNAs, which are more stable and have a longer half-life than non-polyadenylated eRNAs [25]. Production of different isoforms by alternative splicing and cis and trans actions of seRNAs can orchestrate a precise pattern of gene expression [25][26]. As previously mentioned, eRNAs were initially considered as transcriptional noise of enhancers. Later, by using experimental methods, including global run-on sequencing (GRO-seq), Start-seq, and CRISPR/Cas9, several investigations have revealed that a subclass of eRNAs contribute to enhancer function, especially the regulation of gene expression [27][28][29]. As most eRNAs are unstable, their recognition is mainly proceeded via precision nuclear run-on sequencing (PRO-seq), GRO-seq, chromatin immunoprecipitation (ChIP-seq) [30][31][32], or cap analysis of gene expression (CAGE) sequencing [33] rather than the common RNA-seq. Using the CRISPR-Display method, eRNAs were demonstrated to bind to catalytically dead Cas9 (dCas9) for targeting a particular locus of a genome [34]. In another approach, single-molecule fluorescence in situ hybridization (smFISH) [35][36] and ChIRP-seq [37][38] were used as powerful methods for detection of eRNA loci in the genome. Overexpression and knockdown studies of eRNAs demonstrated that this group of non-coding RNAs have strong correlation with their target mRNAs [39]. This correlation is largely dependent on the proximity and correct interactions between the enhancer and promoter. Moreover, chromatin interaction studies revealed that enhancer–promoter looping structure induces higher expression of eRNAs in comparison with other enhancer regions [40][41]. Some studies suggested that eRNAs can act as a cis regulatory element and initiate or stabilize enhancer–promoter looping through association between TFs, mediators, cohesins, and RNApol II [42][43]. Moreover, eRNAs were shown to function in trans for modifying the chromatin structure and directing chromatin accessibility at protein-coding promoter regions [44]. The interaction of eRNAs with CBP and p300 histone acetyltransferases were shown to have a prominent impact on the modulation of H3K27 acetylation and methylation as eRNA knockdown led to decreased levels of H3K27ac and increased levels of H3K27me3 at target-promoter regions [45][46][47][48]. Upon interaction with enhancers, Polycomb repressive complex 1 and 2 (PRC1 and PRC2) have been shown to play regulatory roles in Polycomb-mediated gene transcription [49][50]. Although PRC1 and PRC2 have gene repression activities, in some cases it has been proposed that Polycomb chromatin domains can affect gene expression by forming chromatin topologies that support gene induction [51]. PRC2, for instance, composed of the EZH2 and SUZ12 subunits, which is responsible for establishing and maintaining histone H3K27 methylation during cell differentiation. The interaction of eRNAs and the EZH2 subunit of the PRC2 complex represses its methyltransferase activity and consequently leads to reduced H3K27me3 level and increased gene expression [1][39]. Direct interaction of eRNA with RNApol II, TFs, and general cofactors was shown to be required for initiation and elongation of transcription [14][52]. NELF and P-TEFb complexes are negative and positive elongation factors, respectively, which are released and recruited to RNApol II in the elongation phase. eRNA interacts with NELF and P-TEFb and further promotes the release of paused RNApol II and transition to active elongation by acting as decoys for these complexes [14][47].
Figure 2. Biogenesis of typical eRNA and seRNA and their corresponding function. Active enhancers are bidirectionally transcribed to produce eRNAs and seRNAs. Super enhancers are augmented with higher amount of transcription factors, mediators, and RNApol II compared to enhancers. Therefore, the transcription activity at a super enhancer is typically higher than at a distinct enhancer. From the functional perspective, super enhancers have a greater potential to stimulate target gene transcription.
3. Functional Roles of eRNAs in Cancer
Given that enhancers are recognized to influence the maintenance of different types of cells, it is not unexpected that their malfunction has emerged as a powerful factor behind numerous types of malignancies. Translocation, duplication, insertion, deletion, or point mutation at enhancer regions, and especially transcription factor binding elements [53][54], are frequently observed in cancers [55][56]. One interesting possibility is that these types of mutations make a difference in eRNA expression that eventually drives cancer development. For instance, specific three-stranded nucleic acid organization of the DNA:RNA hybrid and the related non-template single-stranded DNA, known as R-loop, can be shaped at enhancer regions with exceeded eRNA expression levels. Particularly, R-loops are correlated with genomic instability and DNA injury, proposing an association in the initiation and progression of cancer [57]. Moreover, single-stranded DNA (ssDNA) in R-loops may be an off-target for the action of the activation-induced cytidine deaminase (AID) enzyme [58]. Intrinsically, this enzyme is responsible for initiating somatic hypermutation on ssDNA at immunoglobulin (Ig) loci and preferentially alters cytosine to uridine by deamination [59]. AID off-target positions correlate with extremely transcribed enhancers, which promotes genome instability and tumorigenesis [60].
Several studies uncover roles for individual eRNAs in tumorigenesis of many cancer types, including ovarian, breast, prostate, colorectal, and lung adenocarcinomas, showing that their ectopic expression is strongly linked to enhancer dysfunction [61][62][63]. In tumor cells, eRNAs regulate target genes by both cis- and trans-regulatory activities and, hence, play a crucial role in a variety of important signaling cascades [37][64]. For instance, in colorectal cancer, it has been stated that the presence of Colon Cancer-associated Transcript 1 (CCAT1) eRNA was highly correlated with c-Myc overexpression [63]. MYC is accepted as a crucial regulator of cell proliferation and deregulation of this proto-oncogene associated with the development of many cancer types [65]. In a separate study, the knockdown of oncogenic CCAT1 eRNA in squamous cell carcinomas suppressed the SE-associated genes expression required for the propagation and migration of cancer cells [66]. Net1e eRNA, which is located downstream of NET1 proto-oncogene, is a breast cancer specific eRNA and its knockdown by LNA (locked nucleic acids) antisense RNA was shown to strongly reduce cell proliferation in the MCF7 breast cancer cell line [67]. ARIEL in leukemia [68], HPSE in different cancer types [69], and P2RY2 in bladder cancer [70] are other examples of eRNAs targeted by knockdown approaches that may serve as new therapeutic targets for cancer treatment. In breast cancer, 17b-oestradiol (E2)-bound estrogen receptor α (ER- α) could raise the expression of enhancers close to E2-induced coding genes. These differentially expressed eRNAs were demonstrated to elevate the strength of ER-α activated looping of the enhancer–promoter by direct interaction with cohesin. Targeted knockdown of eRNA from corresponding enhancers attenuated cohesion attachment to the ER-α enhancer and consequently reduced enhancer–promoter looping [37]. Wang et al. indicated that WAKMAR2 can be a new candidate eRNA in modulating the microenvironment of invasive breast cancer cells and its downregulation might influence the immune-related genes expression in favor of tumor progression. eRNAs are implicated in various cancer signaling pathways by potentially modifying their target genes, such as immune checkpoints and clinically actionable genes [71]. By successful delineation of basic eRNA mechanisms, including RNA–RNA, RNA–DNA, and RNA–protein interactions, these eRNAs can be considered as new therapeutic targets and will pave the way for eRNA-based cancer diagnostic and therapeutic approaches [72].
4. Data Resources to Explore eRNA in Cancer
As mentioned before, most eRNAs are unstable and non-polyadenylated with low abundance [2]. Thus, they are not easily detectable in routine RNA-sequencing methods, which are based on polyadenylated RNAs. Alternative techniques rely on measuring promising transcripts, such as global run-on sequencing (GRO-Seq) [7], precision run-on nuclear sequencing (PRO-Seq) [73], and cap analysis gene expression (CAGE) [33] to certify that no eRNA is missed. These methodologies are instrumental for the detection of formerly undiscovered eRNAs and active enhancers. For example, the CAGE technique was applied by the FANTOM consortium for profiling the large amounts of transcriptomes of different types of cells, from which 43,011 enhancer elements were revealed to be transcribed to eRNAs [74]. Since the number of detected eRNA transcripts are increased exponentially, comprehensive databases and computational pipelines are highly required to illustrate and consolidate the eRNA expression profiles in normal and cancerous samples. Currently, two types of eRNA data resources were generated. While datasets such as Ensemble (https://www.ensembl.org, accessed on 1 January 2002), ENCODE (https://www.encodeproject.org, accessed on 5 September 2012), FANTOM (http://fantom.gsc.riken.jp/index.html, accessed on 26 March 2014), and the Roadmap Epigenomics Project (http://www.roadmapepigenomics.org, accessed on 13 October 2010) include numerous annotated regulatory elements containing enhancers, other datasets such as The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov, accessed on 26 September 2013) and Genotype-Tissue Expression (GTEx) (https://gtexportal.org/home, accessed on 29 May 2013) have multi-omic data including RNA-seq and survival data from patient samples and the Cancer Cell Line Encyclopedia (CCLE) (https://portals.broadinstitute.org/ccle/about, accessed on 8 May 2019) that apply genomics and sequencing data in ~1000 cancer cell lines for pan-cancer and tumor-specific analysis of eRNAs. These omics data can be downloaded via Xena platform. UCSC Xena cancer browser (https://xena.ucsc.edu, accessed on 22 May 2020) allows biologists to correlate between genomic and/or phenotypic variables with visualizations and analyses. To facilitate research on eRNA, many enhancer pipelines such as SEdb, HACER, RAEdb, HEDD, DiseaseEnhancer, TiED, SEA, and DENdb [75][76][77][78][79][80][81] have been generated. GeneHancer [82] is one of the most common pipelines, which integrates the enhancer annotations from four altered enhancer resources, including Ensembl, FANTOM, VISTA, and ENCODE [10][83][84][85]. Human enhancer RNA Atlas (HeRA) is another data portal that accommodates data from the ENCODE, FANTOM, and GTEx that presents an expression profile and regulatory network of eRNAs in normal human samples [86]. On the contrary, the eRic (eRNA in cancer) database (https://hanlab.uth.edu/eRic, accessed on 8 October 2019) can predict eRNA functions in cancer via collecting eRNA expression profiles, clinical features, target genes, and drug response [67]. By using RNA-seq data from TCGA and GTEx and using CAGE-defined enhancers annotated by FANTOM, Chen et al. developed The Cancer eRNA Atlas (TCeA) data portal, which provides a high-resolution map of eRNA loci. In this map, SE showed discrete loci with sharp eRNA expression peaks. The annotation of SE activities can be used for a broad range of biomedical investigations, such as immunotherapy response and enhanced explanations of cancer phenotypes by resolving intratumoral heterogeneity [87].
5. eRNAs as Prognostic and Diagnostic Biomarker in Cancer
Even though remarkable progress has been made in the field of cancer research, there are still a number of issues that need to be improved, such as delayed diagnosis and poor prognosis. Non-coding RNAs have gained wide consideration in recent years because of their specific expression and functional diversity in a variety of cancers [88]. They play critical roles in various biological pathways and hold great promise in cancer diagnosis and therapy. Clinical trials have also initiated investigating non-coding RNA-based medications as adjuncts to traditional chemotherapeutics [89]. Regarding eRNAs, an increasing number of studies have reported that these non-coding RNAs have amenable prognostic and diagnostic values due to their tumor-specific expression patterns [90][91]. In this section, researchers will review the present findings on eRNAs and their potential prognostic and diagnostic values in cancers. Table 1 summarizes eRNAs and seRNAs as diagnostic and/or prognostic biomarkers in different cancers.
5.1. Head and Neck Squamous Cell Carcinoma
Head and neck cancer is considered one of the most common malignancies in the world, with ~870,000 new cases and ~440,000 deaths in 2020 [92] in which the most common histological subtype of head and neck cancer is head and neck squamous cell carcinoma (HNSCC). Feng et al. showed the role of certified eRNAs as an innovative biomarker in HNSCC. The group indicated the role of eRNA in 500 HNSCC cases by means of an eRNA expression matrix annotated from the TCGA database. Functional enrichment analyses were carried out using Gene Ontology and the Kyoto Encyclopedia of Genes and Genomes (KEGG). Global expression of eRNAs was increased in tumor tissues compared to normal cases; out 369 differentially expressed eRNAs, 330 were upregulated and 39 were downregulated. According to the eRNA expression matrix and survival information, 5 eRNAs were identified with a correlation with the prognosis value in HNSCC cases, which represent an innovative finding in the molecular mechanisms of HNSCC [93]. Gu et al. demonstrated the role of prognosis-related AP001056.1 eRNA in HNSCC. In this research, an incorporated data analysis methodology was developed to recognize major eRNAs in HNSCC. To discover the RNA levels and clinical data from the TCGA project, the interactive web servers, TANRIC (the Atlas of Noncoding RNAs in Cancer) and cBioPortal were applied. From the obtained 5 significant eRNA candidates, AP001056.1 was the most significant survival-associated eRNA in HNSCC with immune-related ICOSLG as its target gene. While strong associations between AP001056.1 and ICOSLG expression were demonstrated in a number of cancers, the most significant effect on overall survival (OS) was observed in HNSCC [94].
Table 1. eRNAs and seRNAs as diagnostic and/or prognostic biomarkers.
| Cancer Type | eRNAs/seRNAs | Deregulation in Cancer | Target Gene/Pathways | Clinical Sample/Number of TCGA Cases | Sample/Model Information | Application | Ref. |
|---|---|---|---|---|---|---|---|
| HNSCC | ENSR00000188847 ENSR00000250663 ENSR00000313345 ENSR00000317887 ENSR00000336429 |
Up | - | 500 TCGA HNSCC samples | Patient sample | Prognosis | [93] |
| AP001056.1 | Down | ICOSLG | 426 TCGA HNSCC samples | Patient sample | Prognosis | [94] | |
| LUAD | TBX5-AS1 | Down | TBX5 | 10 LUAD samples | Patient sample | Prognosis/Diagnosis | [95] |
| 188 functional eRNAs | 129 Up/59 Down | Cell cycle and immune system-related pathways | 80 LUAD samples/481 TCGA LUAD samples | Patient sample | Prognosis | [62] | |
| CRC | CCAT1 CCAT2 |
Up | c-Myc | 150 CRC samples | Patient sample | Prognosis | [96] |
| RP11-569A11.1 | Down | IFIT2 | 39 CRC samples | Patient sample/cell line | Diagnosis | [97] | |
| PVT1 | Down (epigenetic regulation mediated through aberrant methylation in CRC) | Myc | 698 TCGA CRC dataset | Patient sample | Prognosis | [98] | |
| GC | EMX2OS | Up | EMX2 | 375 TCGA GC samples | Patient sample | Prognosis | [99] |
| FALEC | Up | ECM1 | 60 GC samples | Patient sample/cell line | Prognosis | [100] | |
| HPSE | Up | hnRNPU/p300/EGR1/HPSE axis | 90 GC samples | Patient sample/cell line | Prognosis | [69] | |
| CDK6-AS1 | UP (in patients below 60 years) | CDK6 | 407 TCGA GC samples | Patient sample | Prognosis | [101] | |
| WAKMAR2 | Down | TNFAIP3 | 371 TCGA GC samples | Patient sample | Prognosis | [102] | |
| Breast Cancer | SLIT2 | Down | MAPK/c-Fos signaling pathway | 1211 TCGA breast cancer and 12 bone metastases samples | Patient sample/cell line | Prognosis/Bone metastasis | [103] |
| WAKMAR2 | Down | IL27RA RAC2 FABP7 IGLV1-51 IGHA1 IGHD |
1104 TCGA invasive breast cancer samples | Patient sample | Prognosis | [71] | |
| HCC | DCP1A | Up | PRKCD | 1580 TCGA samples together with 1791 target genes | Patient sample | Prognosis | [104] |
| SPRY4-AS1 | Up | SPRY4 | 124 TCGA samples | Patient sample | Prognosis | [105] | |
| AL445524.1 | Up | CD4-CLTA4 related genes | 371 TCGA HCC tumor samples and 54 adjacent normal specimens |
Patient sample | Prognosis | [106] | |
| Brain Cancer | AC003092.1 | Up | TFPI2 | 161 TCGA GBM patients | Patient sample | Prognosis | [107] |
| CYP1B1-AS1 | Up | CYP1B1 | 10,000 TCGA cancer sufferers covering 33 diverse cancer types | Patient sample | Prognosis | [108] | |
| LINC00844 MRPS31P5 CRNDE |
Down Down Up |
PHYHIPL ATP7B and NEK3 IRX5 |
693 TCGA cohorts and 325 cohort in Chinese Glioma Genome Atlas (CGGA)/40 glioma samples | Patient sample | Prognosis/Diagnosis | [109] | |
| ENSR00000210436 ENSR00000249159 ENSR00000195717 ENSR00000195824 ENSR00000094845 ENSR00000283518 ENSR00000094854 ENSR00000031043 ENSR00000031044 ENSR00000260651 ENSR00000146066 ENSR00000301859 ENSR00000213692 ENSR00000326719 ENSR00000134110 ENSR00000134111 ENSR00000134112 ENSR00000013533 ENSR00000013524 ENSR00000082228 ENSR00000048324 ENSR00000082228 ENSR00000048324 |
Association with immune-related dysfunctions in the TME | ADCYAP1R1 FGF13 PSMB8 MAPT BMPR1A DDX17 ELN BMP2 SEMA6C PDIA2 PTPN6 SSTR5 CD4 |
TCGA and CGGA samples | Patient sample/cell line | prognosis | [110] | |
| Prostate Cancer | K-KLK3 | Up | KLK3 | 45 patient samples | Patient sample/cell line | Diagnosis | [111] |
| PARGP1 | Up | AGAP4 | TCGA database | Patient sample | Prognosis | [112] | |
| Bladder Cancer | MARC1 | Up | − | 37 tissues | Patient sample/cell line | Diagnosis | [113] |
| EMP1 | UP | APOLD1 and GPRC5A/ KRAS signaling, etc. | 411 TCGA bladder urothelial carcinoma samples | Patient sample | Prognosis/Bone metastasis prediction | [114] | |
| ESCA | AC007255.1 | Up | PRR15 | 162 ESCA TANRIC database/12 pairs of ESCA tissues and normal tissues | Patient sample | Prognosis | [115] |
| Colon Adenocarcinoma | LINC02257 | Up | DUSP10 | 521 TCGA samples | Patient sample | Prognosis | [116] |
| Ovarian Cancer | FOXP4-AS1 | Down | FOXP4 | 379 TCGA samples/42 patient samples | Patient sample | Prognosis | [117] |
| Thyroid Cancer | NBDY MEG3 AP002358.1 AC141930.1 |
Relation to the prognosis of thyroid cancer patients | - | 510 TCGA samples | Patient sample | Prognosis/Diagnosis | [90] |
| PAAD | LINC00242 | Down | PHF10 | 177 PAAD data set from UCSC | Patient sample/cell line | Prognosis | [91] |
Abbreviations: HNSCC, head and neck squamous cell carcinoma; LUAD, lung adenocarcinoma; CRC, colorectal cancer; GC, gastric cancer; HCC, hepatocellular carcinoma; ESCA, esophageal cancer; PAAD, pancreatic adenocarcinoma.
This entry is adapted from the peer-reviewed paper 10.3390/ncrna8050066
References
- Han, Z.; Li, W. Enhancer RNA: What we know and what we can achieve. Cell Prolif. 2022, 55, e13202.
- Sartorelli, V.; Lauberth, S.M. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 2020, 27, 521–528.
- Chepelev, I.; Wei, G.; Wangsa, D.; Tang, Q.; Zhao, K. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res. 2012, 22, 490–503.
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112.
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936.
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2010, 470, 279–283.
- Wang, D.; Garcia-Bassets, I.; Benner, C.; Li, W.; Su, X.; Zhou, Y.; Qiu, J.; Liu, W.; Kaikkonen, M.; Ohgi, K.A.; et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011, 474, 390–394.
- Wan, L.; Li, W.; Meng, Y.; Hou, Y.; Chen, M.; Xu, B. Inflammatory Immune-Associated eRNA: Mechanisms, Functions and Therapeutic Prospects. Front. Immunol. 2022, 13, 849451.
- Desanta, F.; Barozzi, I.; Mietton, F.; Ghisletti, S.; Polletti, S.; Tusi, B.K.; Muller, H.; Ragoussis, J.; Wei, C.-L.; Natoli, G. A Large Fraction of Extragenic RNA Pol II Transcription Sites Overlap Enhancers. PLoS Biol. 2010, 8, e1000384.
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461.
- Kim, T.-K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187.
- Core, L.J.; Waterfall, J.J.; Gilchrist, D.A.; Fargo, D.C.; Kwak, H.; Adelman, K.; Lis, J.T. Defining the Status of RNA Polymerase at Promoters. Cell Rep. 2012, 2, 1025–1035.
- Kim, Y.W.; Lee, S.; Yun, J.; Kim, A. Chromatin looping and eRNA transcription precede the transcriptional activation of gene in the β-globin locus. Biosci. Rep. 2015, 35, 1–8.
- Schaukowitch, K.; Joo, J.-Y.; Liu, X.; Watts, J.K.; Martinez, C.; Kim, T.-K. Enhancer RNA Facilitates NELF Release from Immediate Early Genes. Mol. Cell 2014, 56, 29–42.
- Arner, E.; Daub, C.O.; Vitting-Seerup, K.; Andersson, R.; Lilje, B.; Drabløs, F.; Lennartsson, A.; Rönnerblad, M.; Hrydziuszko, O.; Vitezic, M.; et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 2015, 347, 1010–1014.
- Tyssowski, K.; DeStefino, N.R.; Cho, J.-H.; Dunn, C.J.; Poston, R.G.; Carty, C.E.; Jones, R.D.; Chang, S.M.; Romeo, P.; Wurzelmann, M.K.; et al. Different Neuronal Activity Patterns Induce Different Gene Expression Programs. Neuron 2018, 98, 530–546.e11.
- Herz, H.-M.; Hu, D.; Shilatifard, A. Enhancer Malfunction in Cancer. Mol. Cell 2014, 53, 859–866.
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108.
- Melo, C.A.; Drost, J.; Wijchers, P.J.; van de Werken, H.; de Wit, E.; Vrielink, J.A.O.; Elkon, R.; Melo, S.A.; Léveillé, N.; Kalluri, R.; et al. eRNAs Are Required for p53-Dependent Enhancer Activity and Gene Transcription. Mol. Cell 2013, 49, 524–535.
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the Enhancer Landscape during Macrophage Activation Is Coupled to Enhancer Transcription. Mol. Cell 2013, 51, 310–325.
- Beckedorff, F.; Blumenthal, E.; Dasilva, L.F.; Aoi, Y.; Cingaram, P.R.; Yue, J.; Zhang, A.; Dokaneheifard, S.; Valencia, M.G.; Gaidosh, G.; et al. The Human Integrator Complex Facilitates Transcriptional Elongation by Endonucleolytic Cleavage of Nascent Transcripts. Cell Rep. 2020, 32, 107917.
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 2013, 153, 307–319.
- Bruter, A.V.; Rodionova, M.D.; Varlamova, E.A.; Shtil, A.A. Super-Enhancers in the Regulation of Gene Transcription: General Aspects and Antitumor Targets. Acta Nat. 2021, 13, 4–15.
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934–947.
- Xiao, S.; Huang, Q.; Ren, H.; Yang, M. The mechanism and function of super enhancer RNA. Genesis 2021, 59, e23422.
- Wu, M.; Shen, J. From super-enhancer non-coding RNA to immune checkpoint: Frameworks to functions. Front. Oncol. 2019, 9, 1307.
- Natoli, G.; Andrau, J.-C. Noncoding Transcription at Enhancers: General Principles and Functional Models. Annu. Rev. Genet. 2012, 46, 1–19.
- Henriques, T.; Scruggs, B.S.; Inouye, M.O.; Muse, G.W.; Williams, L.H.; Burkholder, A.; Lavender, C.; Fargo, D.C.; Adelman, K. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 2018, 32, 26–41.
- Sigova, A.A.; Abraham, B.J.; Ji, X.; Molinie, B.; Hannett, N.M.; Guo, Y.E.; Jangi, M.; Giallourakis, C.C.; Sharp, P.A.; Young, R.A. Transcription factor trapping by RNA in gene regulatory elements. Science 2015, 350, 978–981.
- Wang, J.; Zhao, Y.; Zhou, X.; Hiebert, S.W.; Liu, Q.; Shyr, Y. Nascent RNA sequencing analysis provides insights into enhancer-mediated gene regulation. BMC Genom. 2018, 19, 633.
- Hah, N.; Danko, C.G.; Core, L.; Waterfall, J.J.; Siepel, A.; Lis, J.T.; Kraus, W.L. A Rapid, Extensive, and Transient Transcriptional Response to Estrogen Signaling in Breast Cancer Cells. Cell 2011, 145, 622–634.
- Blinka, S.; Reimer, M.H.; Pulakanti, K.; Pinello, L.; Yuan, G.-C.; Rao, S. Identification of Transcribed Enhancers by Genome-Wide Chromatin Immunoprecipitation Sequencing. Enhanc. RNAs Methods Mol. Biol. 2017, 1468, 91–109.
- Murakawa, Y.; Yoshihara, M.; Kawaji, H.; Nishikawa, M.; Zayed, H.; Suzuki, H.; FANTOM Consortium; Hayashizaki, Y. Enhanced Identification of Transcriptional Enhancers Provides Mechanistic Insights into Diseases. Trends Genet. 2016, 32, 76–88.
- Shechner, D.; Hacisuleyman, E.; Younger, S.T.; Rinn, J.L. Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat. Methods 2015, 12, 664–670.
- Tsai, P.-F.; Dell’Orso, S.; Rodriguez, J.; Vivanco, K.O.; Ko, K.-D.; Jiang, K.; Juan, A.H.; Sarshad, A.A.; Vian, L.; Tran, M.; et al. A Muscle-Specific Enhancer RNA Mediates Cohesin Recruitment and Regulates Transcription In trans. Mol. Cell 2018, 71, 129–141.e8.
- Femino, A.M.; Fay, F.S.; Fogarty, K.; Singer, R.H. Visualization of Single RNA Transcripts in Situ. Science 1998, 280, 585–590.
- Li, W.; Notani, D.; Ma, Q.; Tanasa, B.; Nunez, E.; Chen, A.Y.; Merkurjev, D.; Zhang, J.; Ohgi, K.; Song, X.; et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 2013, 498, 516–520.
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic Maps of Long Noncoding RNA Occupancy Reveal Principles of RNA-Chromatin Interactions. Mol. Cell 2011, 44, 667–678.
- Arnold, P.R.; Wells, A.D.; Li, X.C. Diversity and Emerging Roles of Enhancer RNA in Regulation of Gene Expression and Cell Fate. Front. Cell Dev. Biol. 2020, 7, 377.
- Lin, Y.C.; Benner, C.; Mansson, R.; Heinz, S.; Miyazaki, K.; Miyazaki, M.; Chandra, V.; Bossen, C.; Glass, C.K.; Murre, C. Global changes in the nuclear positioning of genes and intra- and interdomain genomic interactions that orchestrate B cell fate. Nat. Immunol. 2012, 13, 1196–1204.
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109–113.
- Hadjur, S.; Williams, L.M.; Ryan, N.K.; Cobb, B.S.; Sexton, T.; Fraser, P.; Fisher, A.G.; Merkenschlager, M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature 2009, 460, 410–413.
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435.
- Mousavi, K.; Zare, H.; Dell’Orso, S.; Grontved, L.; Gutierrez-Cruz, G.; Derfoul, A.; Hager, G.L.; Sartorelli, V. eRNAs Promote Transcription by Establishing Chromatin Accessibility at Defined Genomic Loci. Mol. Cell 2013, 51, 606–617.
- Pnueli, L.; Rudnizky, S.; Yosefzon, Y.; Melamed, P. RNA transcribed from a distal enhancer is required for activating the chromatin at the promoter of the gonadotropin α-subunit gene. Proc. Natl. Acad. Sci. USA 2015, 112, 4369–4374.
- Liang, J.; Zhou, H.; Gerdt, C.; Tan, M.; Colson, T.; Kaye, K.M.; Kieff, E.; Zhao, B. Epstein–Barr virus super-enhancer eRNAs are essential for MYC oncogene expression and lymphoblast proliferation. Proc. Natl. Acad. Sci. USA 2016, 113, 14121–14126.
- Shii, L.; Song, L.; Maurer, K.; Zhang, Z.; Sullivan, K.E. SERPINB2 is regulated by dynamic interactions with pause-release proteins and enhancer RNAs. Mol. Immunol. 2017, 88, 20–31.
- Bose, D.A.; Donahue, G.; Reinberg, D.; Shiekhattar, R.; Bonasio, R.; Berger, S.L. RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 2017, 168, 135–149.e22.
- Chan, H.L.; Beckedorff, F.; Zhang, Y.; Garcia-Huidobro, J.; Jiang, H.; Colaprico, A.; Bilbao, D.; Figueroa, M.E.; LaCava, J.; Shiekhattar, R.; et al. Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat. Commun. 2018, 9, 1–16.
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Cancer 2016, 16, 803–810.
- Blackledge, N.P.; Klose, R.J. The molecular principles of gene regulation by Polycomb repressive complexes. Nat. Rev. Mol. Cell Biol. 2021, 22, 815–833.
- Zhao, Y.; Wang, L.; Ren, S.; Wang, L.; Blackburn, P.R.; McNulty, M.S.; Gao, X.; Qiao, M.; Vessella, R.L.; Kohli, M.; et al. Activation of P-TEFb by Androgen Receptor-Regulated Enhancer RNAs in Castration-Resistant Prostate Cancer. Cell Rep. 2016, 15, 599–610.
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493.
- Fröhling, S.; Döhner, H. Chromosomal abnormalities in cancer. N. Engl. J. Med. 2008, 359, 722–734.
- Xi, Y.; Shi, J.; Li, W.; Tanaka, K.; Allton, K.L.; Richardson, D.; Li, J.; Franco, H.L.; Nagari, A.; Malladi, V.S.; et al. Histone modification profiling in breast cancer cell lines highlights commonalities and differences among subtypes. BMC Genom. 2018, 19, 1–11.
- Franco, H.; Nagari, A.; Malladi, V.; Li, W.; Xi, Y.; Richardson, D.; Allton, K.; Tanaka, K.; Li, J.; Murakami, S.; et al. Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res. 2017, 28, 159–170.
- Aguilera, A.; García-Muse, T. R Loops: From Transcription Byproducts to Threats to Genome Stability. Mol. Cell 2012, 46, 115–124.
- Meng, F.-L.; Du, Z.; Federation, A.; Hu, J.; Wang, Q.; Kieffer-Kwon, K.-R.; Meyers, R.M.; Amor, C.; Wasserman, C.R.; Neuberg, D.; et al. Convergent Transcription at Intragenic Super-Enhancers Targets AID-Initiated Genomic Instability. Cell 2014, 159, 1538–1548.
- Di Noia, J.M.; Neuberger, M.S. Molecular Mechanisms of Antibody Somatic Hypermutation. Annu. Rev. Biochem. 2007, 76, 1–22.
- Hakim, O.; Resch, W.; Yamane, A.; Klein, I.; Kieffer-Kwon, K.-R.; Jankovic, M.; Oliveira, T.; Bothmer, A.; Voss, T.C.; Ansarah-Sobrinho, C.; et al. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 2012, 484, 69–74.
- Mo, J.; Zhang, L.; Li, H.; Duan, H.; Wang, D.; Zhao, X.; Xie, Y. The enhancer RNA ADCY10P1 is associated with the progression of ovarian cancer. J. Ovarian Res. 2022, 15, 1–10.
- Qin, N.; Ma, Z.; Wang, C.; Zhang, E.; Li, Y.; Huang, M.; Chen, C.; Zhang, C.; Fan, J.; Gu, Y.; et al. Comprehensive characterization of functional eRNAs in lung adenocarcinoma reveals novel regulators and a prognosis-related molecular subtype. Theranostics 2020, 10, 11264–11277.
- McCleland, M.L.; Mesh, K.; Lorenzana, E.; Chopra, V.S.; Segal, E.; Watanabe, C.; Haley, B.; Mayba, O.; Yaylaoglu, M.; Gnad, F.; et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J. Clin. Investig. 2016, 126, 639–652.
- Hsieh, C.-L.; Fei, T.; Chen, Y.; Li, T.; Gao, Y.; Wang, X.; Sun, T.; Sweeney, C.J.; Lee, G.-S.M.; Chen, S.; et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. USA 2014, 111, 7319–7324.
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35.
- Jiang, Y.; Jiang, Y.; Xie, J.; Mayakonda, A.; Hazawa, M.; Chen, L.; Xiao, J.; Li, C.; Huang, M.; Ding, L.; et al. Co-activation of super-enhancer-driven CCAT1 by TP63 and SOX2 promotes squamous cancer progression. Nat. Commun. 2018, 9, 1–13.
- Zhang, Z.; Lee, J.; Ruan, H.; Ye, Y.; Krakowiak, J.; Hu, Q.; Xiang, Y.; Gong, J.; Zhou, B.; Wang, L.; et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat. Commun. 2019, 10, 1–12.
- Tan, S.H.; Leong, W.Z.; Ngoc, P.C.T.; Tan, T.K.; Bertulfo, F.C.; Lim, M.C.; An, O.; Li, Z.; Yeoh, A.E.J.; Fullwood, M.J.; et al. The enhancer RNA ARIEL activates the oncogenic transcriptional program in T-cell acute lymphoblastic leukemia. Blood 2019, 134, 239–251.
- Jiao, W.; Chen, Y.; Song, H.; Li, D.; Mei, H.; Yang, F.; Fang, E.; Wang, X.; Huang, K.; Zheng, L.; et al. HPSE enhancer RNA promotes cancer progression through driving chromatin looping and regulating hnRNPU/p300/EGR1/HPSE axis. Oncogene 2018, 37, 2728–2745.
- Ding, M.; Zhan, H.; Liao, X.; Li, A.; Zhong, Y.; Gao, Q.; Liu, Y.; Huang, W.; Cai, Z. Enhancer RNA - P2RY2e induced by estrogen promotes malignant behaviors of bladder cancer. Int. J. Biol. Sci. 2018, 14, 1268–1276.
- Wang, L.; Liu, J.; Tai, J.; Zhou, N.; Huang, T.; Xue, Y.; Quan, Z. A prospective study revealing the role of an immune-related eRNA, WAKMAR2, in breast cancer. Sci. Rep. 2021, 11, 1–13.
- Lee, J.-H.; Xiong, F.; Li, W. Enhancer RNAs in cancer: Regulation, mechanisms and therapeutic potential. RNA Biol. 2020, 17, 1550–1559.
- Mahat, D.B.; Kwak, H.; Booth, G.T.; Jonkers, I.H.; Danko, C.G.; Patel, R.; Waters, C.T.; Munson, K.; Core, L.J.; Lis, J.T. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc. 2016, 11, 1455–1476.
- Liu, F.J.G. Enhancer-derived RNA: A primer. Genom. Proteom. Bioinform. 2017, 15, 196–200.
- Ashoor, H.; Kleftogiannis, D.; Radovanovic, A.; Bajic, V.B. DENdb: Database of integrated human enhancers. Database 2015, 2015, bav085.
- Zhang, G.; Shiwei, Z.; Zhu, S.; Lan, Y.; Xu, L.; Yuan, H.; Liao, G.; Liu, X.; Zhang, Y.; Xiaoqin, L.; et al. DiseaseEnhancer: A resource of human disease-associated enhancer catalog. Nucleic Acids Res. 2018, 46, D78–D84.
- Xiong, L.; Kang, R.; Ding, R.; Kang, W.; Zhang, Y.; Liu, W.; Huang, Q.; Meng, J.; Guo, Z. Genome-wide Identification and Characterization of Enhancers Across 10 Human Tissues. Int. J. Biol. Sci. 2018, 14, 1321–1332.
- Wang, J.; Dai, X.; Berry, L.D.; Cogan, J.D.; Liu, Q.; Shyr, Y. HACER: An atlas of human active enhancers to interpret regulatory variants. Nucleic Acids Res. 2019, 47, D106–D112.
- Wang, Z.; Zhang, Q.; Zhang, W.; Lin, J.-R.; Cai, Y.; Mitra, J.; Zhang, Z.D. HEDD: Human Enhancer Disease Database. Nucleic Acids Res. 2018, 46, D113–D120.
- Cai, Z.; Cui, Y.; Tan, Z.; Zhang, G.; Tan, Z.; Zhang, X.; Peng, Y. RAEdb: A database of enhancers identified by high-throughput reporter assays. Database 2019, 2019, bay140.
- Wei, Y.; Zhang, S.; Shang, S.; Zhang, B.; Li, S.; Wang, X.; Wang, F.; Su, J.; Wu, Q.; Liu, H.; et al. SEA: A super-enhancer archive. Nucleic Acids Res. 2016, 44, D172–D179.
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Stein, T.I.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028.
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801.
- Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fairley, S.; Fitzgerald, S.; et al. Ensembl 2012. Nucleic Acids Res. 2012, 40, D84–D90.
- Visel, A.; Minovitsky, S.; Dubchak, I.; Pennacchio, L.A. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 2007, 35, D88–D92.
- Zhang, Z.; Hong, W.; Ruan, H.; Jing, Y.; Li, S.; Liu, Y.; Wang, J.; Li, W.; Diao, L.; Han, L. HeRA: An atlas of enhancer RNAs across human tissues. Nucleic Acids Res. 2021, 49, D932–D938.
- Chen, H.; Liang, H. A High-Resolution Map of Human Enhancer RNA Loci Characterizes Super-enhancer Activities in Cancer. Cancer Cell 2020, 38, 701–715.e5.
- Qian, Y.; Shi, L.; Luo, Z. Long Non-coding RNAs in Cancer: Implications for Diagnosis, Prognosis, and Therapy. Front. Med. 2020, 7, 902.
- Le, P.; Romano, G.; Nana-Sinkam, P.; Acunzo, M. Non-Coding RNAs in Cancer Diagnosis and Therapy: Focus on Lung Cancer. Cancers 2021, 13, 1372.
- Liang, Y.; Zhang, Q.; Xin, T.; Zhang, D.-L. A four-enhancer RNA-based prognostic signature for thyroid cancer. Exp. Cell Res. 2022, 412, 113023.
- Tong, W.; Zhu, L.; Bai, Y.; Yang, L.; Liu, Z.; Zhang, Y. Enhancer RNA LINC00242-Induced Expression of PHF10 Drives a Better Prognosis in Pancreatic Adenocarcinoma. Front. Oncol. 2021, 11, 795090.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Feng, G.; Wang, T.; Xue, F.; Qi, Y.; Wang, R.; Yuan, H. Identification of enhancer RNAs for the prognosis of head and neck squamous cell carcinoma. Head Neck 2021, 43, 3820–3831.
- Gu, X.; Wang, L.; Boldrup, L.; Coates, P.J.; Fahraeus, R.; Sgaramella, N.; Wilms, T.; Nylander, K. AP001056.1, A Prognosis-Related Enhancer RNA in Squamous Cell Carcinoma of the Head and Neck. Cancers 2019, 11, 347.
- Cheng, L.; Han, T.; Chen, B.; Nie, K.; Peng, W. TBX5-AS1, an enhancer RNA, is a potential novel prognostic biomarker for lung adenocarcinoma. BMC Cancer 2021, 21, 1–10.
- Thean, L.F.; Blöcker, C.; Li, H.H.; Lo, M.; Wong, M.; Tang, C.L.; Tan, E.K.W.; Rozen, S.G.; Cheah, P.Y. Enhancer-derived long non-coding RNAs CCAT1 and CCAT2 at rs6983267 has limited predictability for early stage colorectal carcinoma metastasis. Sci. Rep. 2021, 11, 1–7.
- Chen, H.; Zheng, J.; Yan, L.; Zhou, X.; Jiang, P.; Yan, F. Super-enhancer–associated long noncoding RNA RP11-569A11.1 inhibited cell progression and metastasis by regulating IFIT2 in colorectal cancer. J. Clin. Lab. Anal. 2021, 35, e23780.
- Shigeyasu, K.; Toden, S.; Ozawa, T.; Matsuyama, T.; Nagasaka, T.; Ishikawa, T.; Sahoo, D.; Ghosh, P.; Uetake, H.; Fujiwara, T.; et al. The PVT1 lncRNA is a novel epigenetic enhancer of MYC, and a promising risk-stratification biomarker in colorectal cancer. Mol. Cancer 2020, 19, 1–6.
- Liu, G.-X.; Tan, Y.-Z.; He, G.-C.; Zhang, Q.-L.; Liu, P. EMX2OS plays a prognosis-associated enhancer RNA role in gastric cancer. Medicine 2021, 100, e27535.
- Wu, H.; Qiao, F.; Zhao, Y.; Wu, S.; Hu, M.; Wu, T.; Huang, F.; Chen, W.; Sun, D.; Liu, M.; et al. Downregulation of Long Non-coding RNA FALEC Inhibits Gastric Cancer Cell Migration and Invasion Through Impairing ECM1 Expression by Exerting Its Enhancer-Like Function. Front. Genet. 2019, 10, 255.
- Yang, S.; Zou, X.; Yang, H.; Li, J.; Zhang, A.; Zhang, L.; Li, C.; Zhu, L.; Ma, Z. Identification of Enhancer RNA CDK6-AS1 as a Potential Novel Prognostic Biomarker in Gastric Cancer. Front. Genet. 2022, 13, 854211.
- Zhang, Y.; Yan, Y.; Ning, N.; Shen, Z.; Ye, Y. WAKMAR2, a Prognosis-related Enhancer RNA in Gastric Cancer. Res. Sq. 2020.
- Li, P.; Lin, Z.; Liu, Q.; Chen, S.; Gao, X.; Guo, W.; Gong, F.; Wei, J.; Lin, H. Enhancer RNA SLIT2 Inhibits Bone Metastasis of Breast Cancer Through Regulating P38 MAPK/c-Fos Signaling Pathway. Front. Oncol. 2021, 11, 4172.
- Wu, H.; Zhang, J.; Bai, Y.; Zhang, S.; Zhang, Z.; Tong, W.; Han, P.; Fu, B.; Zhang, Y.; Shen, Z. DCP1A is an unfavorable prognostic-related enhancer RNA in hepatocellular carcinoma. Aging 2021, 13, 23020–23035.
- Ye, M.; Wang, S.; Qie, J.-B.; Sun, P.-L. SPRY4-AS1, A Novel Enhancer RNA, Is a Potential Novel Prognostic Biomarker and Therapeutic Target for Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 765484.
- Cai, S.; Hu, X.; Chen, R.; Zhang, Y. Identification and Validation of an Immune-Related eRNA Prognostic Signature for Hepatocellular Carcinoma. Front. Genet. 2021, 12, 967.
- Guo, X.-Y.; Zhong, S.; Wang, Z.-N.; Xie, T.; Duan, H.; Zhang, J.-Y.; Zhang, G.-H.; Liang, L.; Cui, R.; Hu, H.-R.; et al. Immunogenomic Profiling Demonstrate AC003092.1 as an Immune-Related eRNA in Glioblastoma Multiforme. Front. Genet. 2021, 12.
- Ye, T.; Li, L.-L.; Peng, X.-M.; Li, Q. CYP1B1-AS1 Is a Novel Biomarker in Glioblastoma by Comprehensive Analysis. Dis. Markers 2021, 2021, 1–8.
- Lin, H.; Yang, Y.; Hou, C.; Zheng, J.; Lv, G.; Mao, R.; Xu, P.; Chen, S.; Zhou, Y. An integrated analysis of enhancer RNAs in glioma and a validation of their prognostic values. Am. J. Transl. Res. 2021, 13, 8611.
- Tian, W.; Chen, K.; Yan, G.; Han, X.; Liu, Y.; Zhang, Q.; Liu, M. A Novel Prognostic Tool for Glioma Based on Enhancer RNA-Regulated Immune Genes. Front. Cell Dev. Biol. 2022, 9, 798445.
- Nishimura, K.; Mori, J.; Sawada, T.; Nomura, S.; Kouzmenko, A.; Yamashita, K.; Kanemoto, Y.; Kurokawa, T.; Hayakawa, A.; Tokiwa, S.; et al. Profiling of Androgen-Dependent Enhancer RNAs Expression in Human Prostate Tumors: Search for Malignancy Transition Markers. Res. Rep. Urol. 2021, 13, 705–713.
- Ang, X.; Xu, Z.; Zhou, Q.; Zhang, Z.; Ma, L.; Zhang, X.; Zhou, F.; Chen, W. PARGP1, a specific enhancer RNA associated with biochemical recurrence of prostate cancer. All Life 2021, 14, 774–781.
- Liu, Y.; Ding, M.; Liao, X.; Gao, Q.; He, A.; Liu, B.; Hu, K.; Xie, H.; Zhou, Q.; Zhan, H.; et al. High expression of enhancer RNA MARC1 or its activation by DHT is associated with the malignant behavior in bladder cancer. Exp. Cell Res. 2018, 370, 303–311.
- Hao, Z.; Yan, P.; Zheng, B.; Li, Z.; Yang, J.; Liu, R.; Gong, B.; Huang, Z.; Zha, Z. Identification of the prognostic Enhancer RNA in bladder cancer bone metastasis. Res. Sq. 2020.
- Wang, Q.; Yu, X.; Yang, N.; Xu, L.; Zhou, Y. LncRNA AC007255.1, an immune-related prognostic enhancer RNA in esophageal cancer. PeerJ 2021, 9, e11698.
- Xiao, J.; Liu, Y.; Yi, J.; Liu, X. LINC02257, an Enhancer RNA of Prognostic Value in Colon Adenocarcinoma, Correlates With Multi-Omics Immunotherapy-Related Analysis in 33 Cancers. Front. Mol. Biosci. 2021, 174, 646786.
- Hua, T.; Tian, Y.-J.; Wang, R.-M.; Zhao, C.-F.; Kong, Y.-H.; Tian, R.-Q.; Wang, W.; Ma, L.-X. FOXP4-AS1 is a favorable prognostic-related enhancer RNA in ovarian cancer. Biosci. Rep. 2021, 41, BSR20204008.
This entry is offline, you can click here to edit this entry!