Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Mitochondria are essential organelles and crucial for cellular survival. Mitochondrial biogenesis and mitophagy are dynamic features that are essential for both maintaining the health of the mitochondrial network and cellular demands. The accumulation of damaged mitochondria has been shown to be related to a wide range of pathologies ranging from neurological to musculoskeletal.
- mitophagy
- mitochondria
- Mitochondrial Homeostasis
1. Introduction
Mitochondria are essential organelles and are responsible for energy production for cellular survival. Therefore, dysfunctional or damaged mitochondria are related to a wide spectrum of pathologies, especially in tissues with the highest energy requirements [1]. Because mitochondria are crucial for the maintenance of the energy level of cells and for cellular survival, mitochondrial homeostasis is strictly regulated by mitochondrial biogenesis and mitophagy [2,3,4]. Mitophagy is the selective elimination process of damaged mitochondria by engulfment, and several molecular pathways are required for properly functioning mitophagy [5].
2. Mitochondrial Homeostasis
Mitochondria are essential organelles in cellular metabolism and physiology. Defective mitochondria are related to a broad spectrum of pathologies ranging from neurological to musculoskeletal [1]. Mitochondrial biogenesis and autophagy of mitochondria are two main pathways that preserve mitochondrial content and quality in cells. The tight regulation of these opposing pathways enables cells to adapt to the cellular energy demand and to respond to both intracellular and extracellular stress [12]. Mitochondria have their own circular genome that encodes 13 essential proteins for respiratory complexes. However, most mitochondrial proteins are encoded by nuclear DNA synthesized within the cytosol and imported into mitochondria. Several transcription factors are activated to control mitochondrial biogenesis in response to a variety of stimuli, including nutrition availability, hormones, growth factors, and temperature changes. Nuclear respiratory factors (NRF1 and NRF2), estrogen-related receptors (ERR-α, ERR-β, and ERR-γ), and the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) are among the key regulators of mitochondrial biogenesis. NRF1 and NRF2 drive the expression of the necessary nuclear genes encoding the proteins related to mitochondrial functioning. Additionally, transcription factor A (TFAM) and transcription factor B (TFB) proteins are important regulators of mitochondrial DNA transcription and replication and are regulated by both NRF1 and NRF2 [3,12,13] (Figure 1). Another group of nuclear hormone receptors including estrogen-related receptors ERR-α, ERR-β, and ERR-γ can stimulate mitochondrial biogenesis. They mainly regulate the transcription of genes involved in Krebs’ cycle, fatty acid oxidation, mitochondrial fusion, and fission. Furthermore, PGC-1α, a member of peroxisome proliferator-activated receptors (PPARs), serves as a coactivator and governs the activity of a wide range of transcription factors related to mitochondrial proliferation [4,12].
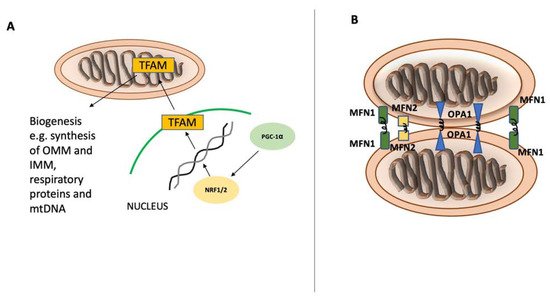
Figure 1. Mitochondrial biogenesis and fusion. Mitochondrial biogenesis and fusion. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is the main regulator of mitochondrial biogenesis (A) and translocates into the nucleus after its activation. In the nucleus, PGC-1α stimulates NRF1 and NRF2 expression, leading to increased activity of mitochondrial transcription factor A (TFAM), which orchestrates the synthesis of mtDNA and proteins. Mitochondrial fusion (B) is controlled by mitofusin-1 (MFN1) and mitofusin-2 (MFN2) on the mitochondrial outer membrane and by optic atrophy 1 (OPA1) on the inner mitochondrial membrane.
As damaged mitochondria can be detrimental to cellular homeostasis, several quality control mechanisms have evolved to ensure the smooth functioning of cells, including the production of adenosine triphosphate (ATP) [14]. Mitochondrial fusion is the joining of two mitochondria through their outer and inner membrane interfaces via membrane GTPases called mitofusin-1 (MFN1), mitofusin-2 (MFN2), and optic atrophy protein-1 (OPA1) (Figure 1). This mechanism allows dysfunctional mitochondria to exchange metabolites and proteins to cope with oxidative stress and enhance their overall respiratory function [15]. However, mitochondrial fusion is a rescue mechanism and is selective for polarized and active mitochondria. If mitochondria are severely damaged or depolarized, they become the target of autophagy for digestion and elimination [16]. Autophagy is an evolutionarily conserved quality control mechanism by which sequestered cytoplasmic components and organelles are packaged in double-membrane vesicles to be transported to the lysosome for degradation. This pathway enables cells to eliminate unwanted, dangerous, and damaged components, including dysfunctional mitochondria. Depending on the component to be degraded, autophagy can be selective or non-selective. In non-selective autophagy, the phagophore sequesters large sections of the cytoplasm for degradation. This kind of autophagy typically occurs during starvation with low nutritional levels. However, selective autophagy is a result of the autophagosome’s evolutionary adaptation to tackle the number and integrity of cellular organelles, such as ER, ribosomes, peroxisomes, and mitochondria. Selective autophagy receptors (SARs) are necessary for the identification of cargo and attachment of the cargo to the phagophore. Protein alterations, primarily phosphorylation, ubiquitination, acetylation, and oligomerization, control the activity of SARs [17,18,19]. Mitophagy is termed as selective destruction of damaged mitochondria, during which a portion of the mitochondrial network is gradually engulfed within a double-membraned autophagosome called mitophagosome. It is a process that involves multiple molecular pathways and is strictly regulated.
In order to facilitate mitophagy, certain prerequisites must be met following mitochondrial damage, such as fission of the mitochondrial network, priming of the mitochondrion for recognition, and engulfment of the mitochondrion via the formation of autophagosome [5]. The GTPase dynamin-related protein 1 (Drp1) catalyzes mitochondrial fission, which involves splitting of the mitochondrial network into smaller pieces so mitochondria can be accommodated in the phagosome. Several adaptor proteins found on the surface of the mitochondria recruit Drp1 from cytosol at locations where endoplasmic reticulum (ER) tubules encounter mitochondria. This is followed by Drp1 oligomerization and GTP hydrolysis, which leads membrane constriction at fission sites [5,20]. It is also essential to understand how mitochondrial fission and mitophagy are coupled. PTEN-induced putative kinase 1 (PINK1), a critical mitophagy-initiating kinase, has been shown to alleviate the inhibitory effects of protein kinase A (PKA) on Drp1 phosphorylation and to promote fission [21]. Another key mitophagy receptor, FUN14 domain-containing 1 (FUNDC1), stimulates fission by direct association with Drp1 [22]. Cells possess multiple mitophagy mechanisms (Figure 2), and various stimuli can promote mitophagy via multiple signaling cascades in different cellular contexts [23].
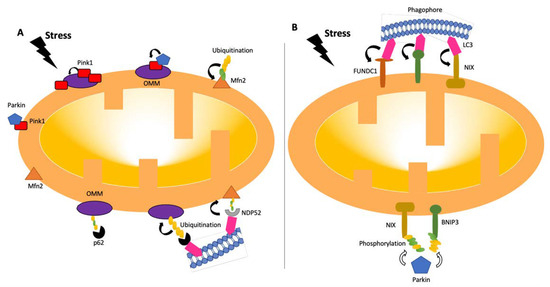
Figure 2. Pathways of mitophagy. Mitophagy pathways are either receptor- (B) or ubiquitin- (A) dependent [24,26]. Under stress conditions, mitochondria become dysfunctional, and PINK1 accumulates and stabilizes on the outer membrane of mitochondria, triggering PARKIN activation [27]. PARKIN recruits the ubiquitination and phosphorylation processes of outer-membrane proteins. Adaptor proteins, such as p62 and NDP52, recognize phosphorylated poly-ubiquitin chains. LC3 connects to adaptor proteins, and phagophores recognize LC3. In the receptor-mediated pathway (B), LC3 attaches to FUNDC1, BNIP3, or NIX receptors. BNIP3 or NIX can also be phosphorylated (PARKIN-mediated or -independent). LC3: microtubule-associated protein 1A/1B-light chain 3; BNIP3: Bcl-2/adenovirus E1B 19-kDa-interacting protein 3; FUNDC1: mitochondrial receptor FUN14 domain-containing 1; NIX: BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like.
At times of mitochondrial malfunctioning, mitophagy eliminates dysfunctional mitochondria and maintains the population in a healthy state. Cells have developed many frequently overlapping processes to ensure that mitophagy can proceed in a balanced manner in response to a variety of stimuli and triggers because the precise and timely removal of mitochondria appears to be vital for cell survival [2]. Mitophagy regulatory pathways can be classified as ubiquitin-dependent or -independent. However, studies have revealed that crosstalk occurs between these signaling pathways [23,24].
In the ubiquitin-dependent pathway, poly-ubiquitin-tagged outer mitochondrial membrane components are required for recognition by autophagic machinery. Among the regulators of ubiquitin-dependent mitophagy are the PINK1-Parkin pathways. These two major proteins play crucial roles in damage-induced mitophagy [25,26,27]. Under stable conditions, PTEN-induced putative kinase 1 (PINK1) is constantly transported from the outer mitochondrial membrane (OMM) to the inner mitochondrial membrane (IMM) and cleaved by several proteases in functional mitochondria. This process allows the cellular levels of PINK1 to remain low [28,29]. Following the incident leading to potential mitochondrial membrane disruption, PINK1 acts as a molecular sensor of mitochondrial damage. The transportation of PINK1 is prevented due to mitochondrial membrane depolarization and causes the accumulation of PINK1 on OMM. Parkin is translocated to the mitochondrial surface as a result of PINK1 stabilization on OMM and its activation by autophosphorylation. The full activation of Parkin by PINK1-mediated phosphorylation at the respective serine 65 residues leads to conformational changes in the intramolecular structure of Parkin. Then, Parkin promotes elongation of pre-existing ubiquitin chains on OMM, which also serve as PINK1 substrates [30,31]. Parkin facilitates a feed-forward mechanism to produce poly-Ub chains, boosting mitophagy signals [2,29,32]. Phosphorylated poly-Ub chains on mitochondrial proteins are recognized by adaptor proteins (p62, OPTN, and NDP52 (nuclear dot protein 52)), which then bind to LC3 to initiate the production of autophagosomes [2]. Along with Parkin, several additional ubiquitin E3 ligases, including Gp78, SMURF1, SIAH1, MUL1, and ARIH1, are involved in the control of mitophagy. After localization on the mitochondrial outer membrane, they produce ubiquitin chains, which cause the recruitment of certain autophagy adaptors, such as optineurin (OPTN), nuclear dot protein 52 (NDP52), and p62, among others [2,33,34]. Once adaptor proteins are attached, they directly interact with light chain 3 (LC3), anchoring ubiquitinated mitochondria to autophagosomes [2].
Besides ubiquitin-dependent pathways, various mitochondrial proteins, including NIP-3 like protein X (NIX), FUNDC1 (FUN14 domain containing 1), and BNIP3 also serve as mitophagy receptors and direct defective mitochondria to autophagosomes for elimination. Those mitophagy receptors directly engage with LC3 through their LC3 interaction region (LIR) motifs and provide another mechanism for the destruction of dysfunctional mitochondria [35]. Whereas NIX plays a crucial role in programmed mitophagy during differentiation, BNIP3 alters the dynamics of the mitochondria, leading to the splitting of damaged mitochondria by causing OPA1 to disassemble and release and DDRP1 to be recruited to the mitochondrial surface [36]. Through control of Parkin recruitment, NIX and BNIP3 maintain mitochondrial homeostasis, indicating crosstalk between mitophagy receptors and the PINK1-Parkin pathway [37].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10102539
This entry is offline, you can click here to edit this entry!