Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Others
Drug delivery systems transfer medications to target locations throughout the body. These systems are often made up of biodegradable and bioabsorbable polymers acting as delivery components. The introduction of density functional theory (DFT) has tremendously aided the application of computational material science in the design and development of drug delivery materials. The use of DFT and other computational approaches avoids time-consuming empirical processes.
- drug delivery system
- DFT
- polymer
- nanocomposites
- quantum mechanics
1. Introduction
One of the most exciting areas of biomedical research is the investigation of innovative drug delivery methods. To improve medication cell/tissue selectivity, rate of release, therapeutic index, and bioavailability, multidisciplinary scientific techniques integrating traditional or engineered technologies are applied. The building elements of these systems are typically biodegradable and bioabsorbable polymers, with their copolymers serving as delivery components [1,2]. To boost the therapeutic efficiency of medications, therapeutic agents, and vaccines, drug delivery systems are used to transport them to a specific location within the body with a regulated release [3]. Computational methodologies, such as DFT, have been developed to bypass time-consuming empirical procedures for the optimization of these formulations.
Density functional theory (DFT) computations, in particular, offer outstanding levels of accuracy with comparable computation time and are more inexpensive in terms of computational resources than other ab initio approaches currently in use. Additionally, it avoids the many-electron wavefunction in favour of electron density and has the potential benefit of dealing with only one function of a single spatial coordinate. Moreover, it employs generalized gradient approximations (GGAs), which use the density gradient to generate a more precise function [4,5,6].
DFT are a strong and low-cost method for revealing a material’s fundamental information, including energy, geometric structure, electrical, and optical characteristics. It offers important theoretical predictions and assistance from the standpoint of material design. It provides crucial information at the levels of atoms, molecules, and unit cells from the perspective of interpreting the results. The influence of element doping on the geometric and electrical characteristics of polymer-based drug carriers, as well as the interaction between the drugs and the nanocarriers, is considerably aided by DFT calculations [7,8].
For example, Kazemi and colleagues used DFT calculations to examine the hydrogen bonding interactions between letrozole and methacrylic acid-trimethylolpropane trimethacrylate copolymers as drug delivery systems [9]. Meanwhile, Karatars and co-workers investigated the interaction of curcumin with a poly(lactic-co-glycolic acid) and montmorillonite composite in a drug delivery system from the first principle calculation [10]. The drug delivery potential of hexagonal boron nitride (h-BN) and PEGylated h-BN (PEG-h-BN) for the delivery of the anticancer medication Gemcitabine (Gem) were investigated by Farzaneh and colleagues using DFT simulations. With adsorption energy of -15.08 kJ/mol, the drug physically adsorbs into the h-BN surface by the growth of π-π stacking. The findings also show that the PEG group grafting to h-BN resulted in π-π stacking, which is enhanced by the generation of strong hydrogen bonds and resulted in a 20% increase in adsorption energy (−90.74 kJ/mol) [11].
2. DFT: A Quick Overview
The first-principles approach is based on quantum mechanics (QM), which expresses the conduction of electrons and atomic nuclei in every condition. The basic equation in this computation is the Schrödinger equation (Equation (1)). The many-body problem occurs in many-electron systems when electrons interact with one another [12,13,14].

where Ψ is the wavefunction, Ĥ is Hamiltonian, and E is the system’s energy.
Several approximations, such as the Born–Oppenheimer approximation, were devised to solve the complex Schrödinger equation. Two theorems proposed by Hohenberg and Kohn served as the foundations of DFT. The Hohenburg–Kohn equation may be written as Equation (2) according to the first theorem.
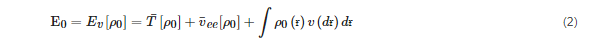
where T¯ is the sum of electronic kinetic energy, ν(ɍ) is the function for nuclear potential energy for an electron at a point ɍ, v¯ is electron–electron repulsion, and the overbars denote the average variables.
The second hypothesis proposes a density minimum principle, stating that the ground state energy of any trial electron density (ρtr) cannot be lower than that of the true ground system and may be represented by Equation (3).

he electronic density n(ɍ) is defined by fulfilling the requirement that n(ɍ) d ɍ is the probability of finding any electron in the volume d3ɍ around ɍ. It is simply |ϕ(ɍ)|2 for a single electron with wavefunction ϕ(ɍ). In DFT, we represent the ground-state energy in terms of n(ɍ) instead of Ψ [15,16].

Moreover, the Kohn–Sham technique is a method for calculating atom and molecule energy, structure, and characteristics [17,18].
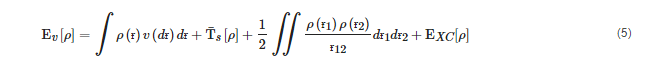
EXC stands for exchange-correlation (XC) energy, which consists of correlation, exchange, columbic correlation, and self-interaction correction. To determine the electronic structural characteristics of polymer-based drug delivery systems, the DFT method has been widely employed. The traditional DFT approach, which uses LDA and GGA exchange-correlation (XC) functionals, produces a band gap that is underestimated. Several approximations such as DFT+U, DMFT, hybrid functionals (B3LYP, PBE0, and HSE), GW approximation, etc., have been proposed to attain better results. Although hybrid functionals and GW give a more exact value of band gap, they come at a hefty computational expense.
The materials science community has access to several DFT codes. The programs are based on various potentials, basis sets, exchange-correlation functionals, and Schrödinger equation-solving techniques. ABINIT, ADF, CASTEP, DMol3, ONETEP, Gaussian, and GAMESS are some examples of DFT programs. Table 1 summarizes the different DFT-based software, the functionals, and the basis set used in polymer-based drug delivery systems.
Table 1. Calculations in polymer-based drug delivery systems using DFT software applications.
| Polymeric Carrier | Drug/Therapeutic Agent | DFT Code | DFT Functional/Basis Set | Ref. |
|---|---|---|---|---|
| Polylactic-co-glycolic acid and montmorillonite | Curcumin | COSMO module of TURBOMOLE V6.1 | B97-D/TZVP | [10] |
| Disulfur-bridged polyethyleneglycol/DOX nanoparticles | Chlorin e6 (Ce6) | Jaguar module in Schrödinger | M06-2x/6-311(d,p) method |
[19] |
| Hyaluronic acid and Zr-based porphyrinic MOF (HA-PCN-224) | Doxorubicin | DMol3 program | PBE/DNP 4.4 | [20] |
| polymer membranes (PDMS, PVA, PUI, PTMG-650-MDI-AP, PPG-725-MDI-AP, PCL-1250-MDI-AP) | Pure liquid solvents | Dmol3 module in Accelrys Materials Studio | GGA/VWN-BP/DNP v4.0.0 | [21] |
| PEG-h-BN | Gemcitabine GEM | GaussView software, Gaussian 09 package | M06e2X/ 6-31G(d,p) |
[11] |
| Methacrylic acid-trimethylolpropane trimethacrylate copolymers | Letrozole | Gaussian-98 | B3LYP and B3PW91 levels/ 6-31G(d) |
[9] |
| Acrylamide-base hydrogel | Antimalarial (chloroquine, primaquine, and amodiaquine) | - | wb97xd/6-31++G(d,p) | [22] |
| Chitosan, silicon dioxide, and graphene oxide | Cisplatin | Gaussian 09 | B3LYP/LANL2DZ | [23] |
| Graphene oxide/polyethylene glycol | Sumatriptan | Gaussian | B3LYP/6-31+G(d) | [24] |
| Polylactic-co-glycolic acid | Anti-cancer sgents of thiazoline | Gaussian 03 | B3LYP/6-31G(d) | [25] |
| Folate functionalized poly(styrene-alt-maleic anhydride), PSMA | Gaussian 09 | B3LYP/6-31G(d,p) | [26] | |
| Polyheterocycles | Antischistosomiasis (praziquantel, niclosamide ethanolamine salt, niclosamide, and trichlorfon) | - | B3LYP | [27] |
| Poly-carboxylic acids functionalized chitosan | Cisplatin | Gaussview 05. | - | [28] |
| PEGylated fluorinated graphene | Camptothecin (CPT) and doxorubicin (DOX) |
- | [29] | |
| Chitosan nanocomposite | Ifosfamide anticancer | COMPASS | - | [30] |
| Polyester dendrimers | Ibuprofen | Gaussian 09 W | M06-2X/6-31G(d) | [31] |
| Polyamidoamine (PAMAM) and polyester dendrimers | Favipiravir | - | M06-2X/6-31G(d) | [32] |
| Hyperbranched polysiloxane containing β-cyclodextrin |
Ibuprofen | - | B3LYP/6-31G(d) | [33] |
| chitosan | Nucleobase (DNA/RNA) | - | B3LYP/6-31++G(d,p) | [34] |
| Poly(O-vinyl carbamate-alt-sulfones) | Mucosal drug (Rhodamine B) | - | B3LYP, 6-311++G- (d,p) |
[35] |
| PAMAM dendrimers | 5-Fluorouracil | Gaussian 09 | B3LYP/6-31G(d,p) and M06-2X/6-31G(d,p) | [36] |
| Chitosan nanoparticle | Hydroxyurea | Gaussian 09 | B3LYP/6-31G(d,p) and M06-2X/6-31G(d,p) | [37] |
| Polyethylene glycol-based nanocomposite | Cephalexin | DMol3 | B3LYP | [4] |
3. Fundamental Properties of Polymer-Based Drug Delivery Systems
DFT computations may be used to derive several characteristics of polymer-based drug delivery systems [11]. To obtain the desired feature under examination, researchers might use a variety of strategies. However, Figure 1 presents a common procedure for applying DFT calculations. The creation of the drug’s and excipient polymer’s molecular structures often marks the beginning of the procedure [38,39]. Additionally, these structures may be imported from both public and commercial molecular structure databases, including the Protein Data Bank [40], PubChem Structure [41,42], and databases for polymer materials. The energy of each particular molecular structure can then be reduced by performing an initial structural relaxation. This enables the drug’s binding energy to be computed when an adsorption calculation is run to determine the most thermodynamically advantageous adsorption sites. On the other hand, interaction energy will be calculated when an adsorption calculation is performed without first optimizing the structure. Ground state calculations are used to further relax the drug-polymer combination that has formed. The generated complex may then be utilized to forecast excited state attributes, such as UV, IR, and using frequency calculations. The optimized complex’s total energy may be determined to examine the thermodynamic properties. The energy gap and molecular orbital characteristics of the drug-polymer configurations can be predicted using electronic simulations in the interim [43]. The many DFT computed characteristics of drug delivery systems based on polymers are explored in the following paragraphs.
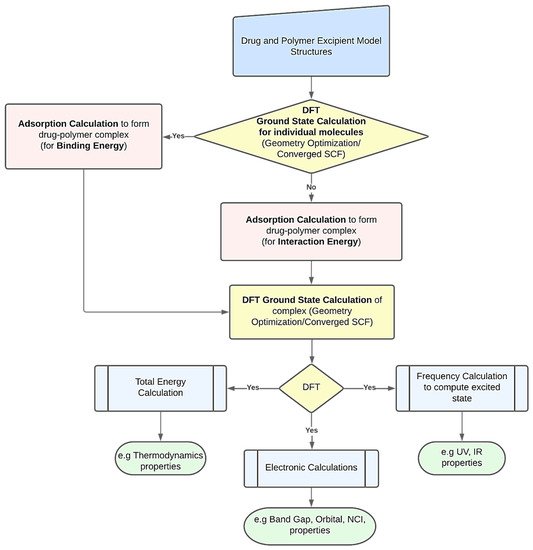
Figure 1. A schematic workflow for employing DFT calculations in polymer-based drug delivery systems.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics14091972
This entry is offline, you can click here to edit this entry!