WRN, the gene responsible for the premature aging associated with Werner syndrome (WS), was identified. Research on genes that suppress aging has been conducted worldwide. During this time, many researchers have participated in these investigations, and it has been established that the protein encoded by WRN is an ATPase activated by single-stranded DNA with DNA unwinding activity in the 3′ to 5′ direction, and that it performs a unique 3′ to 5′ exonuclease activity not observed in other RecQ family members, interacts with a great variety of DNA metabolic proteins, and that it is involved in replication, repair, recombination, transcription, and histone modifications to maintain chromosome stability from the base sequence level to the chromatin level. Within the enzymes/proteins that play crucial roles in these chromosomal events, the WRN helicase plays a fine-tuning role as a supporter. Its functional abnormalities induce chromosomal instability. Abnormal DNA structures accumulated in chromosomes and changes in gene expression profiles caused by epigenetic and transcriptional abnormalities lead to systemic disruption of cellular functions (For example, loss of protein homeostasis, mitochondrial dysfunction, shortened mitotic lifespan, impaired differentiation) and manifest as symptoms such as premature aging.
- premature aging
- RecQ helicases
- senescence
- telomere
- ribosomal DNA
1. Role in Replication Stress Response and DNA Repair
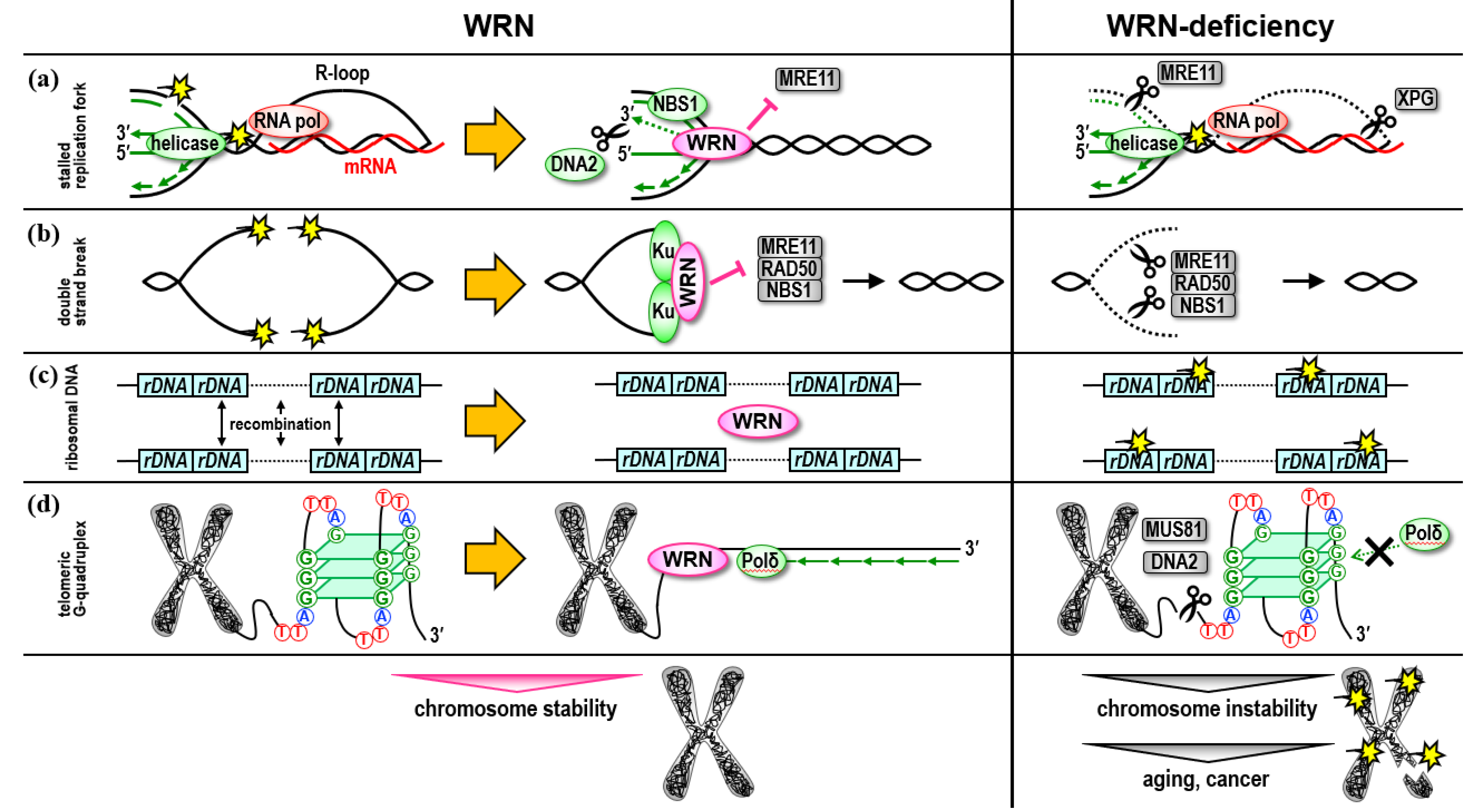
2. Role in the Ribosomal DNA Regions
3. Role in Telomere Maintenance
4. Role in the Regulation of Heterochromatin
5. WRN Helicase as a Molecular Target for Cancer
This entry is adapted from the peer-reviewed paper 10.3390/genes13101802
References
- Sakamoto, S.; Nishikawa, K.; Heo, S.J.; Goto, M.; Furuichi, Y.; Shimamoto, A. Werner Helicase Relocates into Nuclear Foci in Response to DNA Damaging Agents and Co-Localizes with RPA and Rad51. Genes Cells 2001, 6, 421–430.
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 Drives Processing and Restart of Reversed Replication Forks in Human Cells. J. Cell Biol. 2015, 208, 545–562.
- Aiello, F.A.; Palma, A.; Malacaria, E.; Zheng, L.; Campbell, J.L.; Shen, B.; Franchitto, A.; Pichierri, P. RAD51 and Mitotic Function of Mus81 Are Essential for Recovery from Low-Dose of Camptothecin in the Absence of the WRN Exonuclease. Nucleic Acids Res. 2019, 47, 6796–6810.
- Su, F.; Mukherjee, S.; Yang, Y.; Mori, E.; Bhattacharya, S.; Kobayashi, J.; Yannone, S.M.; Chen, D.J.; Asaithamby, A. Nonenzymatic Role for WRN in Preserving Nascent DNA Strands after Replication Stress. Cell Rep. 2014, 9, 1387–1401.
- Lu, H.; Davis, A.J. Human RecQ Helicases in DNA Double-Strand Break Repair. Front. Cell Dev. Biol. 2021, 9, 640755.
- Shamanna, R.A.; Lu, H.; de Freitas, J.K.; Tian, J.; Croteau, D.L.; Bohr, V.A. WRN Regulates Pathway Choice between Classical and Alternative Non-Homologous End Joining. Nat. Commun. 2016, 7, 13785.
- Marabitti, V.; Lillo, G.; Malacaria, E.; Palermo, V.; Sanchez, M.; Pichierri, P.; Franchitto, A. ATM Pathway Activation Limits R-Loop-Associated Genomic Instability in Werner Syndrome Cells. Nucleic Acids Res. 2019, 47, 3485–3502.
- Chappidi, N.; Nascakova, Z.; Boleslavska, B.; Zellweger, R.; Isik, E.; Andrs, M.; Menon, S.; Dobrovolna, J.; Balbo Pogliano, C.; Matos, J.; et al. Fork Cleavage-Religation Cycle and Active Transcription Mediate Replication Restart after Fork Stalling at Co-Transcriptional R-Loops. Mol. Cell 2020, 77, 528–541.e8.
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 Promotes XPG-Mediated R-Loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 2018, 175, 558–570.e11.
- Kobayashi, T. Ribosomal RNA Gene Repeats, Their Stability and Cellular Senescence. Proc. JPN. Acad. Ser. B Phys. Biol. Sci. 2014, 90, 119–129.
- Yamagata, K.; Kato, J.; Shimamoto, A.; Goto, M.; Furuichi, Y.; Ikeda, H. Bloom’s and Werner’s Syndrome Genes Suppress Hyperrecombination in Yeast Sgs1 Mutant: Implication for Genomic Instability in Human Diseases. Proc. Natl. Acad. Sci. USA 1998, 95, 8733–8738.
- Weitao, T.; Budd, M.; Campbell, J.L. Evidence That Yeast SGS1, DNA2, SRS2, and FOB1 Interact to Maintain RDNA Stability. Mutat. Res. 2003, 532, 157–172.
- Marciniak, R.A.; Lombard, D.B.; Johnson, F.B.; Guarente, L. Nucleolar Localization of the Werner Syndrome Protein in Human Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 6887–6892.
- von Kobbe, C.; Bohr, V.A. A Nucleolar Targeting Sequence in the Werner Syndrome Protein Resides within Residues 949-1092. J. Cell Sci. 2002, 115, 3901–3907.
- Karmakar, P.; Bohr, V.A. Cellular Dynamics and Modulation of WRN Protein Is DNA Damage Specific. Mech. Ageing Dev. 2005, 126, 1146–1158.
- Shiratori, M.; Suzuki, T.; Itoh, C.; Goto, M.; Furuichi, Y.; Matsumoto, T. WRN Helicase Accelerates the Transcription of Ribosomal RNA as a Component of an RNA Polymerase I-Associated Complex. Oncogene 2002, 21, 2447–2454.
- Hori, Y.; Shimamoto, A.; Kobayashi, T. The Human Ribosomal DNA Array Is Composed of Highly Homogenized Tandem Clusters. Genome Res. 2021, 31, 1971–1982.
- Faragher, R.G.; Kill, I.R.; Hunter, J.A.; Pope, F.M.; Tannock, C.; Shall, S. The Gene Responsible for Werner Syndrome May Be a Cell Division “Counting” Gene. Proc. Natl. Acad. Sci. USA 1993, 90, 12030–12034.
- Tahara, H.; Tokutake, Y.; Maeda, S.; Kataoka, H.; Watanabe, T.; Satoh, M.; Matsumoto, T.; Sugawara, M.; Ide, T.; Goto, M.; et al. Abnormal Telomere Dynamics of B-Lymphoblastoid Cell Strains from Werner’s Syndrome Patients Transformed by Epstein-Barr Virus. Oncogene 1997, 15, 1911–1920.
- Crabbe, L.; Jauch, A.; Naeger, C.M.; Holtgreve-Grez, H.; Karlseder, J. Telomere Dysfunction as a Cause of Genomic Instability in Werner Syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 2205–2210.
- Wyllie, F.S.; Jones, C.J.; Skinner, J.W.; Haughton, M.F.; Wallis, C.; Wynford-Thomas, D.; Faragher, R.G.; Kipling, D. Telomerase Prevents the Accelerated Cell Ageing of Werner Syndrome Fibroblasts. Nat. Genet. 2000, 24, 16–17.
- Shimamoto, A.; Kagawa, H.; Zensho, K.; Sera, Y.; Kazuki, Y.; Osaki, M.; Oshimura, M.; Ishigaki, Y.; Hamasaki, K.; Kodama, Y.; et al. Reprogramming Suppresses Premature Senescence Phenotypes of Werner Syndrome Cells and Maintains Chromosomal Stability over Long-Term Culture. PLoS ONE 2014, 9, e112900.
- Shimamoto, A.; Yokote, K.; Tahara, H. Werner Syndrome-Specific Induced Pluripotent Stem Cells: Recovery of Telomere Function by Reprogramming. Front. Genet. 2015, 6, 10.
- Gray, M.D.; Shen, J.C.; Kamath-Loeb, A.S.; Blank, A.; Sopher, B.L.; Martin, G.M.; Oshima, J.; Loeb, L.A. The Werner Syndrome Protein Is a DNA Helicase. Nat. Genet. 1997, 17, 100–103.
- Shen, J.C.; Gray, M.D.; Oshima, J.; Loeb, L.A. Characterization of Werner Syndrome Protein DNA Helicase Activity: Directionality, Substrate Dependence and Stimulation by Replication Protein A. Nucleic Acids Res. 1998, 26, 2879–2885.
- Suzuki, N.; Shimamoto, A.; Imamura, O.; Kuromitsu, J.; Kitao, S.; Goto, M.; Furuichi, Y. DNA Helicase Activity in Werner’s Syndrome Gene Product Synthesized in a Baculovirus System. Nucleic Acids Res. 1997, 25, 2973–2978.
- Huang, S.; Li, B.; Gray, M.D.; Oshima, J.; Mian, I.S.; Campisi, J. The Premature Ageing Syndrome Protein, WRN, Is a 3′→5′ Exonuclease. Nat. Genet. 1998, 20, 114–116.
- Mohaghegh, P.; Karow, J.K.; Brosh, R.M.; Bohr, V.A.; Hickson, I.D. The Bloom’s and Werner’s Syndrome Proteins Are DNA Structure-Specific Helicases. Nucleic Acids Res. 2001, 29, 2843–2849.
- Ketkar, A.; Voehler, M.; Mukiza, T.; Eoff, R.L. Residues in the RecQ C-Terminal Domain of the Human Werner Syndrome Helicase Are Involved in Unwinding G-Quadruplex DNA. J. Biol. Chem. 2017, 292, 3154–3163.
- Edwards, D.N.; Machwe, A.; Chen, L.; Bohr, V.A.; Orren, D.K. The DNA Structure and Sequence Preferences of WRN Underlie Its Function in Telomeric Recombination Events. Nat. Commun. 2015, 6, 8331.
- Crabbe, L.; Verdun, R.E.; Haggblom, C.I.; Karlseder, J. Defective Telomere Lagging Strand Synthesis in Cells Lacking WRN Helicase Activity. Science 2004, 306, 1951–1953.
- Li, B.; Reddy, S.; Comai, L. The Werner Syndrome Helicase Coordinates Sequential Strand Displacement and FEN1-Mediated Flap Cleavage during Polymerase δ Elongation. Mol. Cell. Biol. 2017, 37, e00560-16.
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247.
- Sun, L.; Nakajima, S.; Teng, Y.; Chen, H.; Yang, L.; Chen, X.; Gao, B.; Levine, A.S.; Lan, L. WRN Is Recruited to Damaged Telomeres via Its RQC Domain and Tankyrase1-Mediated Poly-ADP-Ribosylation of TRF1. Nucleic Acids Res. 2017, 45, 3844–3859.
- Opresko, P.L.; von Kobbe, C.; Laine, J.-P.; Harrigan, J.; Hickson, I.D.; Bohr, V.A. Telomere-Binding Protein TRF2 Binds to and Stimulates the Werner and Bloom Syndrome Helicases. J. Biol. Chem. 2002, 277, 41110–41119.
- Opresko, P.L.; Mason, P.A.; Podell, E.R.; Lei, M.; Hickson, I.D.; Cech, T.R.; Bohr, V.A. POT1 Stimulates RecQ Helicases WRN and BLM to Unwind Telomeric DNA Substrates. J. Biol. Chem. 2005, 280, 32069–32080.
- Lombard, D.B.; Beard, C.; Johnson, B.; Marciniak, R.A.; Dausman, J.; Bronson, R.; Buhlmann, J.E.; Lipman, R.; Curry, R.; Sharpe, A.; et al. Mutations in the WRN Gene in Mice Accelerate Mortality in a P53-Null Background. Mol. Cell. Biol. 2000, 20, 3286–3291.
- Chang, S.; Multani, A.S.; Cabrera, N.G.; Naylor, M.L.; Laud, P.; Lombard, D.; Pathak, S.; Guarente, L.; DePinho, R.A. Essential Role of Limiting Telomeres in the Pathogenesis of Werner Syndrome. Nat. Genet. 2004, 36, 877–882.
- Lago, S.; Nadai, M.; Cernilogar, F.M.; Kazerani, M.; Domíniguez Moreno, H.; Schotta, G.; Richter, S.N. Promoter G-Quadruplexes and Transcription Factors Cooperate to Shape the Cell Type-Specific Transcriptome. Nat. Commun. 2021, 12, 3885.
- Tian, Y.; Wang, W.; Lautrup, S.; Zhao, H.; Li, X.; Law, P.W.N.; Dinh, N.-D.; Fang, E.F.; Cheung, H.H.; Chan, W.-Y. WRN Promotes Bone Development and Growth by Unwinding SHOX-G-Quadruplexes via Its Helicase Activity in Werner Syndrome. Nat. Commun. 2022, 13, 5456.
- Yokokura, T.; Kamei, H.; Shibano, T.; Yamanaka, D.; Sawada-Yamaguchi, R.; Hakuno, F.; Takahashi, S.-I.; Shimizu, T. The Short-Stature Homeobox-Containing Gene (Shox/SHOX) Is Required for the Regulation of Cell Proliferation and Bone Differentiation in Zebrafish Embryo and Human Mesenchymal Stem Cells. Front. Endocrinol. 2017, 8, 125.
- Parry, A.J.; Narita, M. Old Cells, New Tricks: Chromatin Structure in Senescence. Mamm Genome 2016, 27, 320–331.
- De Cecco, M.; Criscione, S.W.; Peckham, E.J.; Hillenmeyer, S.; Hamm, E.A.; Manivannan, J.; Peterson, A.L.; Kreiling, J.A.; Neretti, N.; Sedivy, J.M. Genomes of Replicatively Senescent Cells Undergo Global Epigenetic Changes Leading to Gene Silencing and Activation of Transposable Elements. Aging Cell 2013, 12, 247–256.
- Swanson, E.C.; Manning, B.; Zhang, H.; Lawrence, J.B. Higher-Order Unfolding of Satellite Heterochromatin Is a Consistent and Early Event in Cell Senescence. J. Cell Biol. 2013, 203, 929–942.
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. Aging Stem Cells. A Werner Syndrome Stem Cell Model Unveils Heterochromatin Alterations as a Driver of Human Aging. Science 2015, 348, 1160–1163.
- Flasch, D.A.; Macia, Á.; Sánchez, L.; Ljungman, M.; Heras, S.R.; García-Pérez, J.L.; Wilson, T.E.; Moran, J.V. Genome-Wide de Novo L1 Retrotransposition Connects Endonuclease Activity with Replication. Cell 2019, 177, 837–851.e28.
- Sultana, T.; van Essen, D.; Siol, O.; Bailly-Bechet, M.; Philippe, C.; Zine El Aabidine, A.; Pioger, L.; Nigumann, P.; Saccani, S.; Andrau, J.-C.; et al. The Landscape of L1 Retrotransposons in the Human Genome Is Shaped by Pre-Insertion Sequence Biases and Post-Insertion Selection. Mol. Cell. 2019, 74, 555–570.e7.
- Wolf, D.; Goff, S.P. Embryonic Stem Cells Use ZFP809 to Silence Retroviral DNAs. Nature 2009, 458, 1201–1204.
- Castro-Diaz, N.; Ecco, G.; Coluccio, A.; Kapopoulou, A.; Yazdanpanah, B.; Friedli, M.; Duc, J.; Jang, S.M.; Turelli, P.; Trono, D. Evolutionally Dynamic L1 Regulation in Embryonic Stem Cells. Genes Dev. 2014, 28, 1397–1409.
- Jacobs, F.M.J.; Greenberg, D.; Nguyen, N.; Haeussler, M.; Ewing, A.D.; Katzman, S.; Paten, B.; Salama, S.R.; Haussler, D. An Evolutionary Arms Race between KRAB Zinc-Finger Genes ZNF91/93 and SVA/L1 Retrotransposons. Nature 2014, 516, 242–245.
- Bulut-Karslioglu, A.; De La Rosa-Velázquez, I.A.; Ramirez, F.; Barenboim, M.; Onishi-Seebacher, M.; Arand, J.; Galán, C.; Winter, G.E.; Engist, B.; Gerle, B.; et al. Suv39h-Dependent H3K9me3 Marks Intact Retrotransposons and Silences LINE Elements in Mouse Embryonic Stem Cells. Mol. Cell 2014, 55, 277–290.
- Kato, M.; Takemoto, K.; Shinkai, Y. A Somatic Role for the Histone Methyltransferase Setdb1 in Endogenous Retrovirus Silencing. Nat. Commun. 2018, 9, 1683.
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 Drives IFN in Senescent Cells and Promotes Age-Associated Inflammation. Nature 2019, 566, 73–78.
- Simon, M.; Van Meter, M.; Ablaeva, J.; Ke, Z.; Gonzalez, R.S.; Taguchi, T.; De Cecco, M.; Leonova, K.I.; Kogan, V.; Helfand, S.L.; et al. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab. 2019, 29, 871–885.e5.
- Vazquez, B.N.; Thackray, J.K.; Simonet, N.G.; Chahar, S.; Kane-Goldsmith, N.; Newkirk, S.J.; Lee, S.; Xing, J.; Verzi, M.P.; An, W.; et al. SIRT7 Mediates L1 Elements Transcriptional Repression and Their Association with the Nuclear Lamina. Nucleic Acids Res. 2019, 47, 7870–7885.
- Fukuda, S.; Varshney, A.; Fowler, B.J.; Wang, S.-B.; Narendran, S.; Ambati, K.; Yasuma, T.; Magagnoli, J.; Leung, H.; Hirahara, S.; et al. Cytoplasmic Synthesis of Endogenous Alu Complementary DNA via Reverse Transcription and Implications in Age-Related Macular Degeneration. Proc. Natl. Acad. Sci. USA 2021, 118, e2022751118.
- Futami, K.; Takagi, M.; Shimamoto, A.; Sugimoto, M.; Furuichi, Y. Increased Chemotherapeutic Activity of Camptothecin in Cancer Cells by SiRNA-Induced Silencing of WRN Helicase. Biol. Pharm. Bull. 2007, 30, 1958–1961.
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of Cancer Therapeutic Targets Using CRISPR-Cas9 Screens. Nature 2019, 568, 511–516.
- Chan, E.M.; Shibue, T.; McFarland, J.M.; Gaeta, B.; Ghandi, M.; Dumont, N.; Gonzalez, A.; McPartlan, J.S.; Li, T.; Zhang, Y.; et al. WRN Helicase Is a Synthetic Lethal Target in Microsatellite Unstable Cancers. Nature 2019, 568, 551–556.
- van Wietmarschen, N.; Sridharan, S.; Nathan, W.J.; Tubbs, A.; Chan, E.M.; Callen, E.; Wu, W.; Belinky, F.; Tripathi, V.; Wong, N.; et al. Repeat Expansions Confer WRN Dependence in Microsatellite-Unstable Cancers. Nature 2020, 586, 292–298.
- Aggarwal, M.; Sommers, J.A.; Shoemaker, R.H.; Brosh, R.M. Inhibition of Helicase Activity by a Small Molecule Impairs Werner Syndrome Helicase (WRN) Function in the Cellular Response to DNA Damage or Replication Stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530.
- Aggarwal, M.; Banerjee, T.; Sommers, J.A.; Brosh, R.M. Targeting an Achilles’ Heel of Cancer with a WRN Helicase Inhibitor. Cell Cycle 2013, 12, 3329–3335.
- Datta, A.; Biswas, K.; Sommers, J.A.; Thompson, H.; Awate, S.; Nicolae, C.M.; Thakar, T.; Moldovan, G.-L.; Shoemaker, R.H.; Sharan, S.K.; et al. WRN Helicase Safeguards Deprotected Replication Forks in BRCA2-Mutated Cancer Cells. Nat. Commun. 2021, 12, 6561.
- Gupta, P.; Majumdar, A.G.; Patro, B.S. Enigmatic Role of WRN-RECQL Helicase in DNA Repair and Its Implications in Cancer. J. Transl. Genet. Genom. 2022, 6, 147–156.
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and Overcoming Resistance to PARP Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791.