Lower respiratory tract invasion by severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) results in widespread damage of type II alveolar and pulmonary capillary endothelial cells, disruption of the alveolar-capillary barrier, activation of coagulation, and diffuse thrombogenesis amplified by recruited monocytes and neutrophils and concurrent hypofibrinolysis. These pathobiological mechanisms are associated with a distinct form of acute respiratory distress syndrome (ARDS), characterized by relatively high lung compliance, progressively worsening hypoxemia, low potential for lung recruitment, hyperperfusion of nonaerated lung tissue, and diffuse small-vessel thrombosis.
- brain trauma
- intracranial hypertension
- neuromonitoring
1. Introduction
Acute respiratory distress syndrome (ARDS) is characterized by high-permeability pulmonary edema, widespread compressive atelectasis, and inflammatory disruption of the alveolar-capillary barrier [1]. Neutralization of pulmonary surfactant by proteinaceous exudate [2] and increased intraparenchymal hydrostatic pressures due to accumulation of extravascular lung water promote collapse of dependent lung units. This results in hypoxemia and hypercapnia secondary to intrapulmonary shunt and constriction of the functional surface area of the respiratory membrane [3-5]. At lung unit level, there is “diffuse alveolar damage,” comprising disruption of the alveolar and endothelial cell lining, hyaline membranes, edema, and inflammatory cell infiltration [6]. The current definition of ARDS [7] is presented in Table 1.
Table 1. Berlin Definition of Acute Respiratory Distress Syndrome.
|
Timing |
Within 1 week of known clinical insult or new or worsening respiratory symptoms |
|
|
Chest imaging |
Bilateral opacities on CXR or CT not fully explained by effusions, lobar/lung collapse, or nodules |
|
|
Origin of edema |
Respiratory failure not fully explained by cardiac failure or fluid overload |
|
|
Oxygenation |
Mild |
200 mm Hg < PaO2/FiO2 ≤ 300 mm Hg with PEEP or CPAP ≥ 5 cm H2O |
|
Moderate |
100 mm Hg < PaO2/FiO2 ≤ 200 mm Hg with PEEP ≤ 5 cm H2O |
|
|
Severe |
PaO2/FiO2 ≤ 100 mm Hg with PEEP ≥ 5 cm H2O |
|
CXR, chest x-ray; CT computed tomography; PaO2/FiO2 partial pressure of arterial oxygen to fraction of inspired oxygen ratio; PEEP positive end-expiratory pressure; CPAP continuous positive airway pressure.
Reproduced in concordance with the Creative Commons Attribution License (CC-BY) from Selickman J, Vrettou CS, Mentzelopoulos SD, Marini JJ. COVID-19-Related ARDS: Key Mechanistic Features and Treatments. J Clin Med. 2022;11(16):4896. doi: 10.3390/jcm11164896.
In ″typical ″ [i.e. pre-coronavirus disease (COVID)-19] ARDS, shunt fraction and physiological dead space rise proportionally to the loss of aerated and perfused (i.e. functional) lung tissue. The ARDS lung is heterogenous with functional units interspersed with non-aerated units. The location and distribution of atelectatic lung units is determined by gravity and the presence of external compression by overlying edematous tissue [9,10].
2. Pathobiology of COVID-19 related ARDS (C-ARDS)
Following spread to the lower respiratory tract, SARS-COV-2 binds via its spike protein to angiotensin converting enzyme 2 (ACE-2) receptors of primarily type II alveolar cells [11,12]. The spike protein is then cleaved by a transmembrane serine protease, thereby facilitating the release of the viral ribonucleoprotein into the cytoplasm [13]. Subsequently, viral replicases promote viral RNA transcription using the host cell’s endoplasmic reticulum [14,15].
Intracellular viral RNA and damage associated molecular patterns trigger the release of interferons and pro-inflammatory cytokines, which mediate leukocyte recruitment [16,17]. The diffuse alveolar epithelial injury may promote a state of disequilibrium between coagulation and fibrinolysis and intra-alveolar hyaline membrane formation hindering gas exchange [14,18,19]. Diffuse alveolar injury is tightly associated with a concurrent, small-vessel, endothelial cell activation and damage due to hypoxia, cytokines, leukocyte infiltration and direct viral infection [14,20,21]. An inflammatory, pulmonary and extrapulmonary endothelial cell injury and microcirculatory dysfunction may ensue and contribute to the development of severe C-ARDS and multi-organ failure [14,20].
Severe COVID-19 / C-ARDS is characterized by widespread, immune cell-mediated, small-vessel thrombosis, termed immunothombosis [14,22]. Underlying mechanisms include: interleukin (IL)-6 release by alveolar epithelial cells [14]; coagulation activation by exposed extracellular matrix [14]; tissue factor expression and release of cytokines and neutrophil extracellular traps (NETs) by activated monocytes and neutrophils [14,22,24,25]; SARS-COV-2 complement activation, augmenting tissue factor expression by neutrophils and differentiation of CD-16+ cytotoxic T-cells [14,25,26]; propagation of the immunothrombotic process through platelet recruitment and activation [22,27]; neutralization of anticoagulants by neutrophil elastase and myeloperoxidase [22]; and increased expression of plasminogen activator inhibitor [14,28].
3. C-ARDS vs. ″typical″ ARDS
In contrast to ″typical″ ARDS, C-ARDS is characterized by extensive micro vascular thrombosis, which may involve more than 25% of the lung parenchyma in more than 50% of the patients with severe disease [29-33]. Additional pathological features of C-ARDS include severe endothelial injury, engorged pulmonary capillaries, and more frequent large-vessel pulmonary emboli compared to ″typical″ ARDS [20,21,35-37]. C-ARDS imaging studies also indicate the presence of dilated pulmonary vessels within opacified (i.e. poorly aerated or non-aerated) lung areas [37,38], as well as widespread perfusion defects in areas of normal radiographic density [39,40]. The regional vasodilation-induced overperfusion of gasless tissue has been associated with a shunt fraction-to-gasless tissue ratio of >2 [41,42], and more frequent severe hypoxemia (i.e. PaO2/FiO2 ≤ 100 mm Hg) at higher levels of tidal compliance (e.g. 50 mL/cmH2O) compared to ″typical″ ARDS [43]. Early-C-ARDS hypoxemia is mainly due to hyperperfusion of non-ventilated lung units, rather than non-cardiogenic pulmonary edema [36,44-47]. Nevertheless, following disease progression, late-phase C-ARDS becomes similar to late-phase ″typical″ ARDS, with respect to low lung compliance and capacity, high physiological dead space, and low potential for recruitment [48]. Table 2 displays a summary comparison of the major characteristics of ″typical″ ARDS and C-ARDS.
Table 2. Comparative presentation of major characteristic features of classical ARDS and C-ARDS.
|
|
Classical ARDS |
C-ARDS |
|
Etiology |
Diverse, pulmonary or extrapulmonary (e.g. bacterial or viral pneumonia, severe trauma, aspiration, sepsis, etc.) |
SARS-COV-2 infection of alveolar type 2 cells (primarily) |
|
Hypoxemia (PaO2/FiO2 ≤300 mmHg at a PEEP level of ≥5 cmH2O) |
Acute onset (e.g. within <48 hours after the clinical insult), or progressive onset (i.e. within 7 days after the clinical insult) |
Progressive onset (i.e. within 7 or more days after the onset of COVID-19 symptoms)* |
|
Lung compliance at hypoxemia onset |
Usually low (e.g. <40 cmH2O/L) |
Usually high (e.g. >40 cmH2O/L) |
|
Recruitment potential |
Low or high, depending on the extent / nature of lung unit involvement and associated atelectasis |
Initially low – may increase with disease progression and development of edema and atelectasis |
|
Functional-to-anatomical shunt ratio / hyperperfusion of gasless tissue * |
Usually 0.5-2.0 / No |
Usually > 2.0 / Yes |
|
Alveolar capillary microthrombosis / new vessel growth |
Present / present |
Diffuse (~9 times more prevalent) / marked (2.7 times higher) |
ARDS, acute respiratory distress syndrome; C-ARDS, coronavirus disease (COVID) 19-related ARDS; SARS-COV-2, severe acute, respiratory syndrome coronavirus 2; PaO2/FiO2, oxygen arterial partial pressure-to-fraction of inspired oxygen fraction ratio; PEEP, positive end-expiratory pressure.
*, May predispose to early, profound hypoxemia and the conceptual risk of pre-intubation, patient self-inflicted lung injury. Reproduced in part, in concordance with the Creative Commons Attribution License (CC-BY) from Selickman J, Vrettou CS, Mentzelopoulos SD, Marini JJ. COVID-19-Related ARDS: Key Mechanistic Features and Treatments. J Clin Med. 2022;11(16):4896. doi: 10.3390/jcm11164896.
4. Ventilatory Settings and Maneuvers in C-ARDS
Mechanical ventilation of the ARDS lungs is invariably linked to increased parenchymal mechanical stress and strain, and the potential for ventilator-induced lung injury (VILI) [49,50]. Mechanistically, stress reflects forces tending to cause and oppose lung parenchymal expansion from the resting state, whereas strain reflects the amount of parenchymal mechanical deformation relative to the resting state [51]. Regarding clinical practice, stress is corresponded to end-inspiratory plateau airway pressure and its subcomponents, i.e. driving pressure and positive end-expiratory pressure (PEEP) [49-51]; in addition, strain has been defined as the tidal volume-to-end-expiratory lung volume ratio [49,50]. Both factors, along with flow and respiratory rate, constitute subcomponents of the mechanical power of ventilation [51,52]. In ARDS non-survivors, the calculated, specific mechanical power (i.e. the power per ventilated lung unit [51]) is approximately tenfold higher relative to healthy subjects [51,52]. Furthermore, mechanical power normalized to respiratory compliance and well-aerated lung tissue is associated with intensive care mortality [53].
Low-stress (i.e. plateau pressure target of <30 cmH2O and driving pressure target of <15 cmH2O) and strain (i.e. tidal volume of 4-8 mL/kg predicted body weight) ventilation has been the mainstay of the management of ARDS over the past 2 decades [54-57]. A PEEP level of >10 cmH2O is also suggested to prevent atelectrauma (i.e. small airway epithelial injury) due to tidal recruitment and derecruitment [56-59]. Furthermore, in cases of hypoxemia despite ventilation optimization, "non-staircase" recruitment maneuvers may also be considered [56,57].
Notably, despite the presence of an extensive evidence-base as regards "typical" ARDS, recently gained knowledge about C-ARDS pathophysiology may point toward the need for further, patient-level individualization of the ventilatory management. For example, patients with the "type L" ARDS phenotype (i.e. relatively high lung compliance, low ventilation-to-perfusion ratio, low lung weight by computerized tomography, and low lung recruitability [60,61]) may tolerate tidal volumes of 7-8 mL/kg-predicted and relatively low PEEP levels (e.g. 8-10 cmH2O), without appreciable risk of strain-related, or expiratory derecruitment-related VILI [51,59,63]. In such patients, a higher PEEP level (e.g. ≥14 cmH2O) may reduce lung compliance, and increase lung stress and physiological dead space [51,64,65].
The regional net effect of PEEP depends on the balance between 1) recruitment of previously collapsed and/or poorly aerated lung units (representing increase in functional parenchyma); and 2) overdistention of previously normally aerated lung units (representing potential loss of functional lung tissue and increase in dead space ventilation). Regional overdistention predisposes to hypercapnia and a decreased cardiac output. The latter may be associated with reduced shunt flow and increased PaO2/FiO2; in such cases, the changes in oxygenation do not reflect changes in lung recruitment [66,67].
The predominantly vascular pathophysiology of C-ARDS may explain recent computerized tomographic findings of low recruitability of functional lung units at higher PEEP levels [68,69]. Higher PEEP levels in C-ARDS have also been associated with worsening of gas exchange and respiratory mechanics, suggesting that PEEP-induced overdistention may prevail over PEEP-associated recruitment [64,68,70-72]. These data indicate that PEEP titration should be patient-specific. PEEP level selection could be guided by measurements of respiratory compliance and PaCO2, as their concurrent improvement suggests recruitment of functional lung units. Additional useful bedside "markers"/tests may include the ventilatory ratio [65] and the recruitment to inflation ratio [73].
In "typical," severe ARDS, prone positioning may improve gas exchange, and reduce lung stress / strain, physiological dead space and 28-day / 90-day mortality [74,75]. Relative to the supine position, pronation results in improved shape-matching between the lungs and the chest wall [76,77] (Figure 1). Prone position facilitates recruitment of the "massive" dorsal lung through relief of the superimposed hydrostatic pressure and external compression by the heart and abdominal contents [78,79]. Prone position is also associated with more homogenous distribution of pleural pressure, regional distending forces, and regional lung volumes and gas / tissue ratios [78,80-86]. Furthermore, as the pattern of pulmonary perfusion is minimally affected by body posture, the increased homogeneity of ventilation following pronation is frequently associated with improved ventilation-perfusion matching [85,87-89].
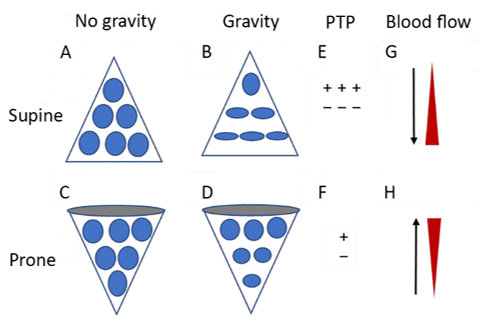
Figure 1. Diagrammatic presentation of physiological mechanisms associated with pronation in the acute respiratory distress syndrome (ARDS). (A) and (C) show the shape of lung units (i.e. alveoli) without the effect of gravity. (B) In the supine position, the volume of dorsal lung units is significantly smaller than the volume of ventral lung units, as a result of gravity and pleural pressure; thus, ventral lung units are more prone to overdistention and dorsal lung units are more prone to compression atelectasis. (D) In the prone position, gravity and pleural pressure result in a decrease in the volume of the ventral lung units and an increase in the volume of the dorsal lung units. (E) In the supine position, the ventral transpulmonary pressure (PTP) may substantially exceed the dorsal PTP. (F) Prone positioning reduces the ventral-to-dorsal PTP gradient, thereby augmenting the homogeneity of ventilation. (G) The reopening, dorsal lung units continue to receive most of the blood flow. (H) The ventral lung units may exhibit a greater tendency to collapse, but are still relatively underperfused. Reproduced in concordance with the Creative Commons Attribution License (CC-BY) from reference 77.
Prone positioning has been reportedly used in approximately 80% of patients with severe C-ARDS [90]. In a recent physiological study of moderate-to-severe C-ARDS (PaO2/FiO2 ≤ 150 mm Hg) [7,91], pronation was associated with a global net recruitment of 6%, a lower concavity index of the time-impedance curve (implying less risk for atelectrauma), and a lower dead space-to shunt ratio (suggesting improved ventilation perfusion-matching). In a multicenter cohort study of C-ARDS, pronation within 48 hours of intensive care unit (ICU) admission was associated with a 16% reduction in the adjusted risk of mortality [92].
Awake pronation relieves dorsal lung compression by the abdomen and mediastinal structures, improves secretion clearance and regional diaphragm movement, and increases functional residual capacity and regional ventilation of dependent lung units [93,94]. In a large meta-trial of severe COVID-19 requiring support with high-flow nasal cannula, awake pronation resulted in a 25% reduction in the risk of tracheal intubation [95]; survival without intubation was higher in patients who tolerated awake pronation for >8 h / day compared to those who did not [96]. Awake pronation may attenuate the risk of patient self-inflicted lung injury by decreasing the inspiratory effort [94,97-99].
5. Non-Ventilatory Therapeutic Interventions in C-ARDS
Several pharmaceutical interventions, including the antiviral remdesivir [100,101], and anti-inflammatory agents such as dexamethasone [102,103], tocilizumab [104], baricitinib [105-107], and anakinra [108] have been associated with improved outcomes of patients with severe COVID-19 / C-ARDS. Extracorporeal membrane oxygenation (ECMO) alone [109] or combined with prone positioning [110-113] may also be considered in patients with severe C-ARDS. Nevertheless, additional high-quality evidence is required to clearly establish the clinical benefit of ECMO in severe C-ARDS. A summary presentation of evidence-based C-ARDS treatment options is provided in Table 3.
Table 3. Evidence-based treatments for coronavirus disease-19 (COVID-19) related acute respiratory distress syndrome (ARDS).
|
Intervention |
Mechanism of Action |
Evidence for Efficacy |
|
Remdesivir day 1: 200 mg IV days 2-10: 100 mg IV |
Inhibition of the viral RNA-dependent, RNA polymerase |
Shortens the time to recovery in hospitalized COVID-19 patients |
|
Dexamethasone days 1-10*: 6 mg IV |
Anti-inflammatory - linked to the activation of the glucocorticoid receptor |
Reduces the probability of in-hospital death in critically ill COVID-19 patients |
|
Tocilizumab single dose: 8 mg/kg IV (max. 800 mg) |
Interleukin-6 antagonism |
Reduces the probability of in-hospital death in critically ill COVID-19 patients |
|
Baracitinib days 1-14*: 4mg† oral or enteral |
Janus kinase inhibition |
Reduces the probability of in-hospital death in critically ill COVID-19 patients |
|
Anakinra days 1-10*: 100 mg subcutaneously |
Interleukin 1 alpha/beta antagonism |
Reduces the probability of in-hospital death in critically ill COVID-19 patients |
|
Prone positioning for at least 16 hours per day until PaO2/FiO2 ≥150 mmHg at PEEP ≤10 cmH2O and FiO2 ≤0.6 |
Attenuation of lung stress and strain Reversal of compression atelectasis Increased homogeneity of ventilation Improved ventilation/perfusion matching |
Reduces the probability of in-hospital death in moderate to severe ARDS |
|
Extracorporeal membrane oxygenation |
Minimization of lung stress and strain ("lung rest") with very low tidal volumes and ventilation pressures |
Possible mortality benefit in severe ARDS |
PaO2/FiO2, oxygen arterial partial pressure-to-inspired oxygen fraction ratio; PEEP, positive end-expiratory pressure.
Reproduced in concordance with the Creative Commons Attribution License (CC-BY) from Selickman J, Vrettou CS, Mentzelopoulos SD, Marini JJ. COVID-19-Related ARDS: Key Mechanistic Features and Treatments. J Clin Med. 2022;11(16):4896. doi: 10.3390/jcm11164896.
6. Conclusions
Key characteristics of early C-ARDS comprise widespread pulmonary (and extrapulmonary) endothelitis and microcirculatory immunothrombosis, in conjunction with hyperperfusion of gasless lung tissue, initial relative preservation of lung compliance with concurrent frequently severe hypoxemia, and low potential for recruitment of functional lung units. Late-phase C-ARDS is associated with low lung compliance, high physiological deadspace and low lung recruitability. Such characteristics may necessitate tailoring of the ventilatory settings to the individual patient, while still avoiding any potentially injurious lung stress and strain. In this context, early pronation and use of relatively low PEEP levels (e.g. 8-10 cmH2O) may be considered. Treatments of proven efficacy should be used, whereas future research should further elucidate the role of ECMO with or without concurrent pronation.
This entry is adapted from the peer-reviewed paper 10.3390/jcm11164896