Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Kidney transplantation is the better choice for patients with end stage kidney disease (ESKD), since it improves the quality of life of the recipients and reduces the associated cost of dialysis. However, long-term transplant survival remains a challenge in the renal transplantation community. The main causes of long-term graft loss are acute and chronic rejection, as well as the complications related to immunosuppression therapy.
- graft rejection
- immune response
- cellular response
- kidney transplantation
1. HLA Antigens
The main antigens recognised by the recipient’s immune system are the components of the highly polymorphic HLA system of the donor [1]. There are two classes of HLA molecules, HLA-I and HLA-II, which are involved in antigen presentation. Moreover, there are three types of HLA class I molecules (HLA-A, -B and -C) and three types of HLA class II molecules (HLA-DP, -DQ and -DR). To date, more than 6000 class I proteins and more than 2000 class II proteins have been found [2]. HLA-I molecules are expressed on all nucleated cells in the body and present endogenous antigens to cytotoxic CD8+ T cells. HLA-II molecules are primarily expressed on antigen-presenting cells (APCs), which are primarily dendritic cells, macrophages and B cells that are involved in the presentation of exogenous antigens to CD 4+ lymphocytes. However, other cell types such as endothelial cells express HLA class II in response to stress or inflammation in response to TNF-α. The host immune system recognises these alloantigens and generates an immune response by generating specific antibodies against the donor HLA system [3].
For organ allocation, a correct HLA donor–recipient match is desirable, as the extent of mismatches at these loci is associated with poorer graft performance. The consequence of an HLA mismatch is the potential generation of antibodies against the mismatched HLA antigen. There is a strong correlation between HLA mismatches at the HLA-A, -B and -DR loci and poor graft outcomes. Compatibility at these loci is still considered the gold standard by the majority of allocation agencies [4].
Although, in the new era of potent immunosuppressants, HLA mismatches seem to have become less important in short-term transplant failure, some studies link the increased number of mismatches to episodes of early subclinical inflammation that would result in poorer long-term transplant outcomes [5].
2. Non-HLA Antigens
There is a growing acknowledgment of the role of other non-HLA antigens in the acute and chronic rejection process. The strongest evidence for the involvement of non-HLA antibodies in graft rejection was provided by reports of antibody-mediated rejection (AMR) in HLA-identical grafts [6][7]. These non-HLA antigens can be polymorphic, such as MICA, or non-polymorphic. The latter are graft-specific antigens that are exposed to the recipient’s immune system in situations of tissue damage and inflammation, such as those caused by ischaemia reperfusion and transplant rejection, and can lead to the formation of autoantibodies [8].
In recent years, an increasing number of non-HLA antigens have been reported to be associated with graft rejection and transplant loss, including angiotensin II type 1 receptor (AT1R), collagen V, perlecan, K-α-tubulin, vimentin and endothelial cell antigens [9].
3. Innate Immunity
Over the past decades, evidence has accumulated, indicating that innate immunity contributes significantly to amplifying and modulating the antigen-specific adaptive immune response against the graft. This makes innate immunity a good therapeutic target to limit the effects of adaptive immunity in transplant rejection [10].
The innate immune system is comprised of cellular components, such as macrophages, neutrophils, dendritic cells and natural killer cells, and blood proteins such as the complement and blood coagulation systems. These three components of innate immunity are closely interrelated and can activate each other. In the early post-transplant period, activation and proinflammatory crosstalk between elements of the innate immune system occur in response to the release of danger-associated molecular patterns (DAMPs) and tissue factor expression in graft cells because of tissue damage and hypoxia following brain death and ischaemia reperfusion. As a result, there will be a potent secretion of proinflammatory cytokines such as IL-6 and IL-21, as well as the activation and recruitment of innate immune cells into the graft [11].
The presence of proinflammatory cytokines in the microenvironment in which T cells recognise the antigen will direct their phenotypic differentiation into tissue-destroying effector T-helper cells (Th1, Th2 and Th17) and limit the differentiation of naïve T cells into tissue-protective regulatory T cells (Tregs) [12]. The latter are immunosuppressive cells that play a central role in immune tolerance and homeostasis. The function of Tregs is involved in the prevention of autoimmune diseases and in the generation of graft tolerance by suppressing the function and proliferation of other CD4+ T cells, CD8+ T cells, B cells, NK cells, macrophages or dendritic cells. Thus, Tregs modulate both the adaptive and innate immunity and are key regulators of inflammation. In organ transplantation, the explosion of proinflammatory cytokines generated by innate immunity leads to an aggressive adaptive response against the graft due to events such as an imbalance between effector T cells and reg T cells [13].
4. T-Cell Allorecognition Pathways
Rejection is caused by an activation of T cells that will develop a coordinated immune response between the adaptive immune response and the innate immune response against the graft. The T cell recognises donor antigens via antigen-presenting cells (APCs) through interactions between the T-cell receptor (TCR) and HLA molecules expressed on APC membranes. In transplantation immunology, both donor and recipient APCs can present antigens to T cells in what is called the direct and indirect allorecognition pathways, respectively [14].
Brain death and ischaemia reperfusion processes produce a secretion of cytokines and DAMPs [15] that will lead to the recruitment and maturation of donor macrophages and dendritic cells in the kidneys, which can then leave to target the recipient’s secondary lymphoid organs and activate the recipient’s T cells via the direct pathway. This direct activation (Figure 1) is a process unique to alloimmunity and occurs by the recognition of intact HLA molecules from donor APCs by recipient’s CD4+ and CD8+ lymphocytes. This pathway is considered to be short-lived due to the short lifespan of the donor APCs, produces a more potent alloresponse than conventional T-cell activation and is considered to be the predominant pathway in the acute cellular rejection process [16].
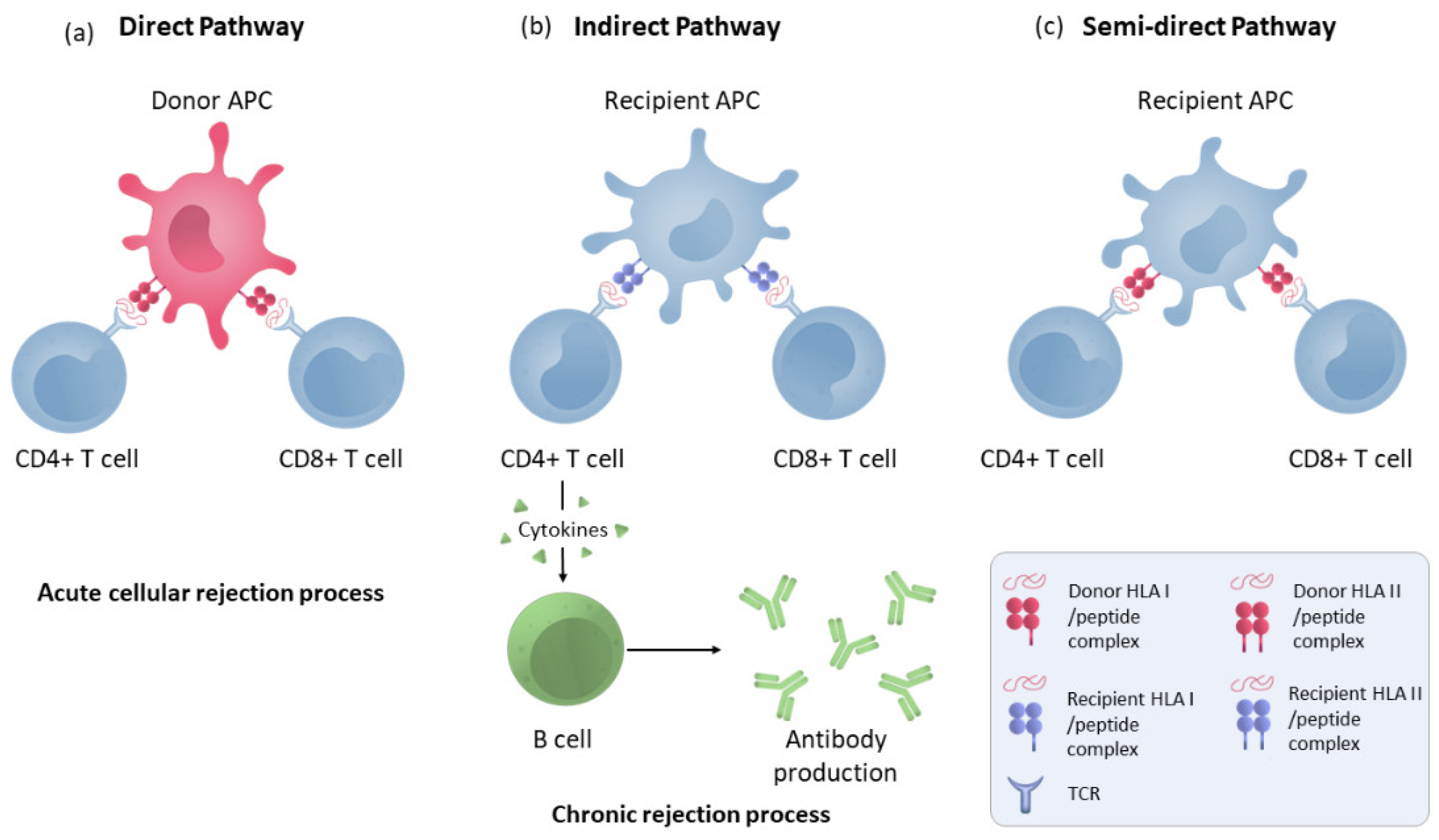
Figure 1. Direct, indirect and semi-direct allorecognition pathways. (a) T-lymphocyte direct pathway activation by HLA molecule presentation by donor APCs to the recipient’s T cells. (b) Indirect pathway recognition by T CD4 and CD8 cells in which the alloantigens presented as peptides processed by recipient APCs through self-HLA molecules. CD4+ lymphocytes activate B cells to make antibodies. (c) T-cell semi-direct pathway activation: T cells recognise intact HLA molecules from the donor, but their own APC presents them.
In the indirect pathway (Figure 1), T CD4 cells recognise the alloantigens presented as peptides processed by recipient APCs trough self-HLA II molecules. The indirect pathway is considered to be long-lasting and predominant in chronic rejection [17]. Indirectly activated CD4 T cells can activate cytotoxic CD8 lymphocytes and are the only cells that can activate alloreactive B cells [18]. Moreover, a semi-direct pathway (Figure 1) has also been described in which T cells recognise intact HLA molecules from the donor, similar to direct recognition, but on their own APC membranes that have acquired these HLA molecules from the donor via various pathways such as cell–cell contacts or exosomes, suggesting that both direct and indirect recognition play a role in chronic rejection [19].
5. Endothelial Cells
The endothelial cells (EC) of the graft will play a fundamental role in the rejection process, as they will be directly exposed to the bloodstream and therefore represent the first barrier between the recipient’s immune system and the graft. EC-expressed antigens are the main targets of the antibodies generated by the recipient’s immune system against the graft. In addition, EC are semi-professional presenting cells that can activate both CD4 and CD8 T-lymphocytes [20].
Under conditions of stress or inflammation, such as those occurring in a transplanted organ, endothelial cells increase the expression of both class I and class II HLA molecules and may be recognised by circulating T lymphocytes. In addition, under these inflammatory conditions, ECs express other minor histocompatibility complex molecules, such as HLA-E and HLA-G, as well as Major Histocompatibility Complex Class I-Related Chain A Antigens (MICA) [21]. MICA is a highly polymorphic protein that has been associated with rejection and late renal failure [22]. Studies have reported that both pre-existing and de novo generated anti-MICA antibodies are associated with acute and chronic rejection [23]. However, MICA is not expressed on resting T or B cells, and therefore, the complement-dependent cytotoxicity crossmatch assay (CDC) test is not valid for detecting anti-MICA antibodies. The MICA polymorphism is in addition to the HLA polymorphism, so it is important to perform donor–recipient matching on the MICA genes, which is currently not done. In addition, MIC molecules function as ligands for NKG2D and can stimulate γδ T and CD8+T cells.
Furthermore, in transplantation, the immune system may react against nonpolymorphic graft antigens, predominantly from endothelial cells, generated as self-antigens in a process involving a loss of B-cell tolerance associated with chronic rejection. In rejection episodes, there is a release of self-antigens in the noncanonical tertiary lymphoid tissues formed in the graft. The cytokine microenvironment of these tissues, enriched in IL-17 and the B-lymphocyte stimulator (BLyS), together with a low concentration of Treg lymphocytes, causes autoreactive B cells to miss deletion signals and differentiate into autoantibody-producing plasma cells [24].
6. T-Cells Activation
Three signals between the T cell and the APC are required for successful T-cell activation. The interaction between the TCR of the lymphocyte and the HLA II molecule with the peptide antigen of the APC results in the first activation signal. It is activated through the calcineurin pathway, which promotes the expression of several cytokines and receptors that are important for lymphocyte proliferation. Lymphocyte activation requires a non-antigen-specific costimulatory signal that, if absent, leads the lymphocyte into a state of anergy and apoptosis. This costimulatory signal requires the interaction of CD28 expressed on the lymphocyte membrane with its ligands, B7.1 (CD80) and B7.2 (CD86), expressed on APCs [25]. Once the T cell has received the activation signal and the costimulatory signal, it will receive further instructions in the form of cytokines, which will induce cell proliferation through IL-2, and define the T-cell subtype into which it will differentiate.
Once activated, a fraction of T cells will differentiate into long-lived memory T cells, which upon re-encounter with the antigen produce an immune response faster and with less need for co-stimulation than naive T cells, and are less susceptible to immunosuppressive treatments [26]. In addition, it is known that memory T cells initially primed by various infectious agents or environmental antigens can cross-react with foreign HLA molecules without having had prior contact with them through a mechanism known as heterologous immunity [27].
The process of T-lymphocyte activation is a common target of the currently used immunosuppressive agents. Immunosuppressive agents such as anti-IL-2 receptor antibodies and the mammalian target of rapamycin (mTor) act by inhibiting the effects of IL-2 [28]. Both cyclosporine and tacrolimus inhibit the calcineurin pathway by binding to their cytoplasmic receptors. Although calcineurin inhibitors (CNIs) have produced an important advance in terms of transplant survival, they produce important side effects due to their nephrotoxicity, and in the long term, they can produce interstitial fibrosis [25].
At the time of transplantation, patients are usually treated with induction therapies directed against T-lymphocytes, such as a T-lymphocyte-depleting agent (thymoglobulin) or IL-2 inhibitors (basilixumab). This has led to a significant reduction in cellular rejection, which is more likely to occur in the first weeks to months post-transplant. It can also occur later, especially in association with reduced immunosuppression [29]. Chronic cellular rejection can present as tubulointerstitial or vascular and is characterised by an infiltration of T-lymphocytes, monocytes and plasma cells into the interstitium, causing tubulitis, or into the subendothelium and intima of the graft arteries, respectively. It is common for cellular and antibody-mediated rejection to coexist in a graft injury. In fact, episodes of early cellular rejection may result in the production of de novo anti-DSA (donor-specific antibodies) antibodies, preceding AMR [30]. However, modern immunosuppressive treatments do not sufficiently address the humoral immune response of the recipient [31]. AMR is responsible for 30–50% of acute rejection cases and 60% of late graft failure [32].
7. B Cells and Humoral Response
B cells play a very important role in the immune response against transplantation, as they are the cells that will produce antibodies against donor-specific antigens that will result in acute or chronic AMR. Once they recognise the antigen via the BCR receptor, B cells internalise the antigen by receptor-mediated endocytosis and migrate to the lymphoid organ, where they present the antigen as a processed peptide via their own HLA II to a cognate Tfh lymphocyte with the involvement of the CD40–CD40L interaction [33]. A fraction of B cells will give rise to short-lived IgM-producing plasma cells, while another fraction of B cells develops in germinal centres, undergoing immunoglobulin class switching, somatic hypermutation and affinity maturation. Once activated, B cells leave the germinal centres as memory B cells or plasma cells. While activated B cells with the highest affinity for antigen differentiate into antibody-producing plasma cells, B cells with less affinity are selected to differentiate into memory B cells. This less stringent selection of memory B cells results in a more diverse repertoire of memory B cells than of antibodies in serum [34]. Plasma cells migrate and settle in the bone marrow or mucosal tissues. These cells are differentiated, nondividing cells that are the main source of antibodies in the serum. Memory B cells remain in quiescent mode and circulate between secondary lymphoid organs and peripheral blood until they encounter the antigen again. After re-encountering the antigen, the memory B cell generates a rapid, enhanced response and differentiates into an antibody-producing cell. Importantly, memory B cells can also be activated in a non-antigen-specific manner through the cytokine environment via bystander activation [35]. In addition, activated B cells are efficient APCs that can interact with naïve T cells and activate them through the HLA-II they express. This makes B cells also mediate and regulate cellular rejection.
Anti-donor antibodies produced by mature B cells are responsible for AMR and can cause a graft injury through several mechanisms. Complement-binding antibodies, the IgG1 and IgG3 subclasses, are capable of activating the complement system, leading to the C5b–C9 membrane attack complex, causing lysis of the target cell. This pathway is detected by the presence in biopsies of C4d, a by-product of the classical complement pathway. In addition, the crystalline fragment (Fc) of both complement-binding and non-complement-binding antibodies acts as a stimulus for FcγR-expressing cells of innate immunity, such as macrophages, dendritic cells and especially natural killer cells, leading to antibody-dependent cell-mediated cytotoxicity [36]. Lastly, the binding of antibodies to HLA molecules on endothelial cells results in a series of endothelial and smooth muscle cell modifications, such as altered cytoskeleton, cell proliferation and the expression of leucocyte adhesion molecules, which generate the histological features of chronic rejection [37].
Acute AMR occurs from the first weeks to years after transplantation. It is characterised by cell death, loss of vascular integrity and tissue damage. Chronic AMR is characterised by a long and progressive process of loss of graft function whose histological features include transplant glomerulopathy, the deposition of fibrillar material in the subendothelium and C4d deposition. For a proper diagnosis of both acute and chronic AMR, according to the criteria of Banff classification, it is necessary to observe by biopsy the pathophysiology associated with each rejection and to detect donor-specific antibodies in the serum. Since the 2013 Banff classification meeting, the presence of C4d in biopsies is not a prerequisite for the diagnosis of AMR but rather an indication of a recent interaction between the antibody and the microvasculature of the endothelium [38]. The treatment of AMR is less effective, and unfortunately, there are no standardised treatments against antibody production.
This entry is adapted from the peer-reviewed paper 10.3390/immuno2040035
References
- Lee, P.C.; Ozawa, M.; Hung, C.J.; Lin, Y.J.; Chang, S.S.; Chou, T.C. Eighteen-Year Follow-Up of a Retrospective Study of HLA Antibody on Kidney Graft Survival. Transplant. Proc. 2009, 41, 121–123.
- Robinson, J.; Mistry, K.; McWilliam, H.; Lopez, R.; Parham, P.; Marsh, S.G.E. The IMGT/HLA database. Nucleic Acids Res. 2011, 39, 1171–1176.
- Thomas, K.A.; Valenzuela, N.M.; Reed, E.F. The perfect storm: HLA antibodies, complement, FcγRs, and endothelium in transplant rejection. Trends Mol. Med. 2015, 21, 319–329.
- Tambur, A.R.; Kosmoliaptsis, V.; Claas, F.H.; Mannon, R.B.; Nickerson, P.; Naesens, M. Significance of HLA-DQ in kidney transplantation: Time to reevaluate human leukocyte antigen matching priorities to improve transplant outcomes? An expert review and recommendations. Kidney Int. 2021, 100, 1012–1022.
- Hernández, D.; Vázquez, T.; Alonso-titos, J.; León, M.; Caballero, A.; Cobo, M.A.; Sola, E.; López, V.; Ruiz-esteban, P.; Cruzado, J.M.; et al. Impact of hla mismatching on early subclinical inflammation in low-immunological-risk kidney transplant recipients. J. Clin. Med. 2021, 10, 1934.
- Collins, A.B.; Chicano, S.L.; Cornell, L.D.; Tolkoff-Rubin, N.; Goes, N.B.; Saidman, S.L.; Farrell, M.L.; Cosimi, A.B.; Colvin, R.B. Putative Antibody-Mediated Rejection With C4d Deposition in HLA-Identical, ABO-Compatible Renal Allografts. Transplant. Proc. 2006, 38, 3427–3429.
- Grafft, C.A.; Cornell, L.D.; Gloor, J.M.; Cosio, F.G.; Gandhi, M.J.; Dean, P.G.; Stegall, M.D.; Amer, H. Antibody-mediated rejection following transplantation from an HLA-identical sibling. Nephrol. Dial. Transplant. 2010, 25, 307–310.
- Zhang, Q.; Reed, E.F. The importance of non-HLA antibodies in transplantation. Nat. Rev. Nephrol. 2016, 12, 484–495.
- Zhang, X.; Reinsmoen, N.L. Impact and production of Non-HLA-specific antibodies in solid organ transplantation. Int. J. Immunogenet. 2020, 47, 235–242.
- Stallone, G.; Pontrelli, P.; Rascio, F.; Castellano, G.; Gesualdo, L.; Grandaliano, G. Coagulation and Fibrinolysis in Kidney Graft Rejection. Front. Immunol. 2020, 11, 1807.
- Pontrelli, P.; Grandaliano, G.; Van Kooten, C. Editorial: Kidney Transplantation and Innate Immunity. Front. Immunol. 2020, 11, 603982.
- Otterbein, L.E.; Fan, Z.; Koulmanda, M.; Thronley, T.; Strom, T.B. Innate immunity for better or worse govern the allograft response. Curr. Opin. Organ Transplant. 2015, 20, 8–12.
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders—Advances and challenges. Hum. Vaccines Immunother. 2022, 18, 2035117.
- Siu, J.H.Y.; Surendrakumar, V.; Richards, J.A.; Pettigrew, G.J. T cell allorecognition pathways in solid organ transplantation. Front. Immunol. 2018, 9, 2548.
- Al-Lamki, R.S.; Bradley, J.R.; Pober, J.S. Endothelial Cells in Allograft Rejection. Transplantation 2008, 86, 1340–1348.
- Ingulli, E. Mechanism of cellular rejection in transplantation. Pediatr. Nephrol. 2010, 25, 61–74.
- Baker, R.J.; Hernandez-Fuentes, M.P.; Brookes, P.A.; Chaudhry, A.N.; Cook, H.T.; Lechler, R.I. Loss of Direct and Maintenance of Indirect Alloresponses in Renal Allograft Recipients: Implications for the Pathogenesis of Chronic Allograft Nephropathy. J. Immunol. 2001, 167, 7199–7206.
- Conlon, T.M.; Saeb-Parsy, K.; Cole, J.L.; Motallebzadeh, R.; Qureshi, M.S.; Rehakova, S.; Negus, M.C.; Callaghan, C.J.; Bolton, E.M.; Bradley, J.A.; et al. Germinal Center Alloantibody Responses Are Mediated Exclusively by Indirect-Pathway CD4 T Follicular Helper Cells. J. Immunol. 2012, 188, 2643–2652.
- Karahan, G.E.; Claas, F.H.J.; Heidt, S. Pre-existing Alloreactive T and B Cells and Their Possible Relevance for Pre-transplant Risk Estimation in Kidney Transplant Recipients. Front. Med. 2020, 7, 340.
- Wang, S.; Zhang, C.; Wang, J.; Yang, C.; Xu, M.; Rong, R.; Zhu, T.; Zhu, D. Endothelial Cells in Antibody-Mediated Rejection of Kidney Transplantation: Pathogenesis Mechanisms and Therapeutic Implications. J. Immunol. Res. 2017, 2017, 8746303.
- Gavlovsky, P.J.; Tonnerre, P.; Guitton, C.; Charreau, B. Expression of MHC class I-related molecules MICA, HLA-E and EPCR shape endothelial cells with unique functions in innate and adaptive immunity. Hum. Immunol. 2016, 77, 1084–1091.
- Luo, L.; Li, Z.; Wu, W.; Luo, G.; Mei, H.; Sun, Z.; Xu, C. The effect of MICA antigens on kidney transplantation outcomes. Immunol. Lett. 2013, 156, 54–58.
- Stastny, P.; Zou, Y.; Fan, Y.; Qin, Z.; Lavingia, B. The emerging issue of MICA antibodies: Antibodies to MICA and other antigens of endothelial cells. Contrib. Nephrol. 2009, 162, 99–106.
- Thaunat, O.; Graff-Dubois, S.; Fabien, N.; Duthey, A.; Attuil-Audenis, V.; Nicoletti, A.; Patey, N.; Morelon, E. A stepwise breakdown of B-cell tolerance occurs within renal allografts during chronic rejection. Kidney Int. 2012, 81, 207–219.
- Lim, M.A.; Kohli, J.; Bloom, R.D. Immunosuppression for kidney transplantation: Where are we now and where are we going? Transplant. Rev. 2017, 31, 10–17.
- Yang, J.; Brook, M.O.; Carvalho-Gaspar, M.; Zhang, J.; Ramon, H.E.; Sayegh, M.H.; Wood, K.J.; Turka, L.A.; Jones, N.D. Allograft rejection mediated by memory T cells is resistant to regulation. Proc. Natl. Acad. Sci. USA 2007, 104, 19954–19959.
- Adams, A.B.; Williams, M.A.; Jones, T.R.; Shirasugi, N.; Durham, M.M.; Kaech, S.M.; Wherry, E.J.; Onami, T.; Lanier, J.G.; Kokko, K.E.; et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J. Clin. Investig. 2003, 111, 1887–1895.
- Goldfarb, D.A. Immunosuppressive Drugs for Kidney Transplantation. J. Urol. 2005, 173, 2105.
- Hara, S. Current pathological perspectives on chronic rejection in renal allografts. Clin. Exp. Nephrol. 2017, 21, 943–951.
- Tsuji, T.; Iwasaki, S.; Makita, K.; Imamoto, T.; Ishidate, N.; Mitsuke, A.; Fukuzawa, N.; Harada, H.; Fukazawa, Y. Preceding T-Cell-Mediated Rejection Is Associated with the Development of Chronic Active Antibody-Mediated Rejection by de Novo Donor-Specific Antibody. Nephron 2021, 144, 13–17.
- Gupta, G.; Abu Jawdeh, B.G.; Racusen, L.C.; Bhasin, B.; Arend, L.J.; Trollinger, B.; Kraus, E.; Rabb, H.; Zachary, A.A.; Montgomery, R.A.; et al. Late antibody-mediated rejection in renal allografts: Outcome after conventional and novel therapies. Transplantation 2014, 97, 1240–1246.
- Voora, S.; Adey, D.B. Management of Kidney Transplant Recipients by General Nephrologists: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 73, 866–879.
- Noelle, R.J.; Snow, E.C. Cognate interactions between helper T cells and B Cells. Immunol. Today 1990, 11, 361–368.
- Lavinder, J.J.; Wine, Y.; Giesecke, C.; Ippolito, G.C.; Horton, A.P.; Lungu, O.I.; Hoi, K.H.; DeKosky, B.J.; Murrin, E.M.; Wirth, M.M.; et al. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 2259–2264.
- Bernasconi, N.L.; Traggiai, E.; Lanzavecchia, A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 2002, 298, 2199–2202.
- Resch, T.; Fabritius, C.; Ebner, S.; Ritschl, P.; Kotsch, K. The Role of Natural Killer Cells in Humoral Rejection. Transplantation 2015, 99, 1335–1340.
- Valenzuela, N.M.; Reed, E.F. Antibodies to HLA Molecules Mimic Agonistic Stimulation to Trigger Vascular Cell Changes and Induce Allograft Injury HHS Public Access. Curr. Transpl. Rep. 2015, 2, 222–232.
- Loupy, A.; Mengel, M.; Haas, M. Thirty years of the International Banff Classification for Allograft Pathology: The past, present, and future of kidney transplant diagnostics. Kidney Int. 2022, 101, 678–691.
This entry is offline, you can click here to edit this entry!