Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The basic composition of the mitogen-activated protein kinase (MAPK) pathway is divided into three modules in sequence, with a cascade effect: MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK), and MAPK. The MAPK signaling pathway is activated in over 50% of human oral cancer cases.
- oral squamous cell carcinoma
- MAPK
- signaling pathway
- immunotherapy
1. Activation of MAPK Signaling Pathway
MAPK activation requires double phosphorylation on the Thr-X-Tyr (X representing any amino acid) motif catalyzed by MAP2K. Upon activation, MAPK phosphorylates specific serine and threonine residues on target substrates, including other protein kinases and many transcription factors. Common and bispecific phosphatases inactivate MAPKs and are regulated by scaffold proteins [1]. Figure 1 demonstrates the cascade activation of the MAPK signaling pathway.
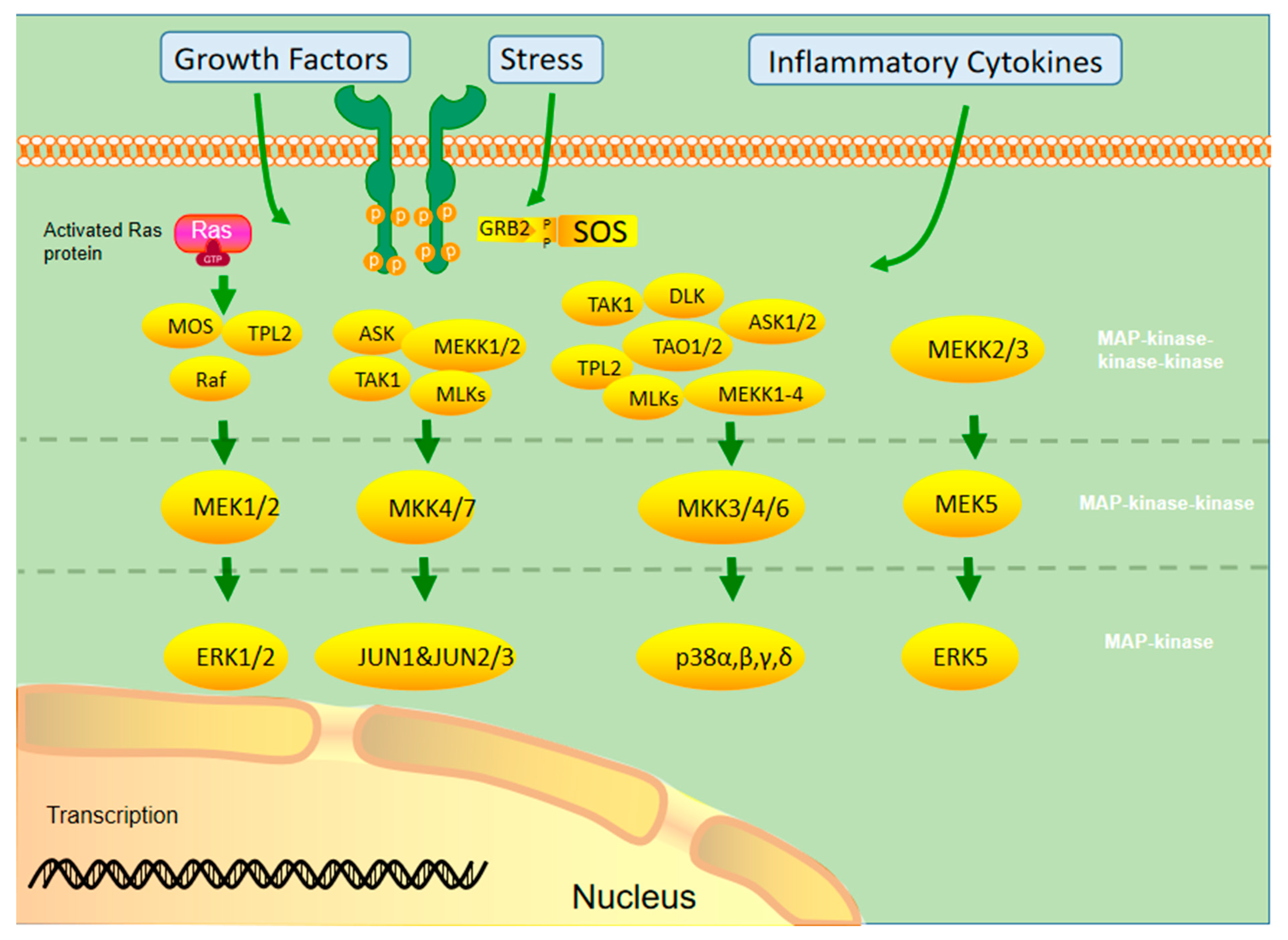
Figure 1. Different MAPKs associate with specific MAPK kinase (MAPKK) and MAPK kinase kinase (MAPKKK) to form a conserved three-stage enzymatic cascade (MAPKKK→MAPKK→MAPK), through which upstream signals are transmitted from MAPK to downstream nuclear transcription factors and cytoskeletal proteins to form a complete MAPK signaling pathway, which finally completes the regulation of cellular physiological activities.
Currently, the ERK1/2 pathway is the most widely studied pathway in the MAPK family. It involves extracellular growth factors, such as the epidermal growth factor (EGF), and activated tyrosine kinase receptors, such as the EGF receptor (EGFR), providing a binding site for the adaptor protein growth factor receptor-bound protein 2 and recruiting the SOS protein into the cell membrane. SOS activates Ras by consuming GTP to form Ras GTP. After Ras activation, it can affect many downstream proteins, including AF6, phosphatidylinositol-3-kinase (PI3K), phospholipase C, and Raf. Among them, Ras GTP recruits the Raf protein into the plasma membrane and phosphorylates it using other kinases (protein kinase A, p21-activated kinase, Src) to activate its kinase function. The Raf protein family includes B-Raf, A-Raf, and C-Raf (Raf1). B-Raf plays an important role in the development and progression of malignant tumors. The activated Raf kinase binds to the downstream MEK 1/2 and activates ERK1/2. Activated ERK1/2 can continue to phosphorylate transcription factors, such as ELK1, ETS, FOS, Jun, myc, and Sp1, and induce gene expression related to cell cycle and cell proliferation. In addition, activated ERK1/2 can phosphorylate various intracellular kinases, such as Raks, msks, and mnks, which affect cell proliferation and adhesion [2][3][4].
The JNK/MAPK signaling pathway can activate cytokines (tumor necrosis factor-alpha [TNF-α], interleukin [IL]-1), EGF, and some G protein-coupled receptors by generating stress through, for example, ultraviolet light, heat shock, hyperosmotic stimuli, and protein synthesis inhibitors. The stress response signal is transmitted to MAPKKK through the Rho subfamily (Rac, rho, Cdc42), a member of the small molecule G protein Ras superfamily, which in turn activates mek4/7 and JNK. JNK phosphorylation can act on various downstream transcription factors (e.g., Jun, ELK1, Ets2, etc.) and kinases (mainly MNK) and produce various physiological processes that promote cell growth, differentiation, survival, and apoptosis [5].
The main inducing factors of the p38/MAPK pathway are hypoxia, ultraviolet radiation, osmotic shock, inflammation, and other stress reactions. P38 MAPK is mainly activated by ERK3/6 phosphorylation, which further promotes cell apoptosis and inhibits cell proliferation by inducing transcription factors and kinases. In addition, this pathway also promotes cell movement [6]. Ras plays an important role in activating the MAPK signal pathway. It is a key component of many cellular signal transduction pathways. Permanent activation of the Ras protein caused by mutation is prevalent in all human cancers. Therefore, Ras inhibitors are effective drugs to treat OSCC [7].
2. Mutation of the MAPK Signaling Pathway
As a signal medium in cells, MAPK controls cell differentiation, proliferation, apoptosis, and other effector functions under the stimulation of external pressure or ligands. The mutation rate of MAPK1 is higher in Asian populations, with an average of 0.79% among 32 cancer genomic profiles from The Cancer Genome Atlas (TCGA) database. The mutation rate of MAPK1 is relatively higher in HNSCC than in pan-cancer TCGA [8][9]. One-fifth of the patients with HNSCC are affected by MAPK pathway mutations, and abnormalities in the MAPK pathway are correlated with the survival time of patients [10]. Chan et al. analyzed the gene-chip technology and showed that MAPK in human HNSCC was overexpressed compared to matched non-cancerous tissues. The expression of several genes in MAPK signaling pathways, such as p38β, ERK2, and JNK2, increased two-folds, showing statistical significance. p38β, JNK2, and ERK2 showed a 5.27-, 2.57-, and 3.30-time increase, respectively [11]. An immunohistochemical analysis of 100s of HNSCC tissues showed elevated active phosphorylated p38 in 79% of the tissues, with increased phosphorylation activities of ERK1/2 and JNK in <33% and <16% of cases, respectively [10][12].
3. MAPK Signaling Pathway in OSCC
3.1. ERK/MAPK Signaling Pathway in OSCC
Activation of ERK1/2 is mostly associated with cell survival, while that of JNK or p38 is associated with the induction of apoptosis [13]. However, this classification is too simplistic, and the actual role of each MAPK cascade depends highly on the cell type and the situation [14]. Activation of MAPK, particularly ERK, is differentially regulated according to the stage of tumor differentiation. For instance, the phosphorylation level of ERK is lower in advanced poorly differentiated prostate cancer than in early prostate cancer [15]. Dickkopf-related protein 3 (Dkk3), as a member of the Dickkopf WNT signaling pathway inhibitor family, has a tumor suppressive effect [16][17]. For example, overexpression of Dkk3 messenger (mRNA) is related to a good prognosis of prostatic cancer [18]. It may induce cancer cell apoptosis by overexpressing through adenovirus-mediated gene transfer [19][20]. Dkk3 plays a carcinogenic role in OSCC. Its overexpression in OSCC cells would largely increase the malignancy of the cells in vitro and in vivo and regulate the malignant behavior of cancer cells through the PI3K/mammalian target of rapamycin (mTOR)/Akt and MAPK pathways. Thus, MAPK may have a tumor- or tissue-specific effects [21]. C-Myc can alter the biological behavior of tumors through the ERK/MAPK pathway. For example, Marconi et al. showed that KRAS mutations activate the Raf/MEK/ERK signaling pathway to upregulate c-Myc, causing overexpressions of Bcl-2, hypoxia-inducible factor (HIF)-1α, vascular endothelial growth factor (VEGF), MMP-9, and other proteins, affecting the invasive, hypoxic, angiogenic, migratory, and inflammatory processes in OSCC [22].
ERK1 and ERK2 are widely expressed in tissues and participate in regulating meiosis, mitosis, and post-mitotic function of differentiated cells. In the early 1980s, ERK1 was the first MAPK core molecule identified in mammals [23]. ERK activation causes phosphorylation and activation of various cytoplasmic substrates, such as cytoskeletal proteins and downstream protein kinases. In addition, phosphorylated ERK1/2 can be transported to the nucleus to activate various transcription factors, such as ELK-1, SP-1, and AP-1, thereby regulating the transcription of different genes [24]. The ERK1/2 signaling pathway mainly affects tumor cells by proliferating cell cycle regulation. Sustained ERK activation can induce cell cycle inhibition and pro-differentiation signals in epithelial origin cells [25]. G1/S conversion is the key regulatory point of the cell cycle. Sustained activation and nuclear localization of ERK1/2 may affect G1/S conversion by regulating cyclin D1 transcription [26][27]. Inhibiting the ERK/MAPK signaling pathway leads to the proliferation, invasion, and migration of OSCC cells, causing G0/G1 arrest and promoting apoptosis [28][29]. Wu et al. [30]. reported that downregulations of MAPK/ERK1/2 and PI3K/Akt signals and cyclin D1 and E expression levels can induce G0/G1 arrest and inhibit OSCC cell proliferation. ERK1/2 is closely associated with tumor invasion and migration, and phosphorylation of ERK/MAPK activates AP-1 and nuclear factor kappa B (NF-κB). They upregulated expressions of MMP2 and MMP9, which are extracellular membrane-degrading enzymes associated with tumor aggressiveness. MMP2 and MMP9 degrade type IV collagen, a major extracellular membrane component of the basement membrane, which may be critical for tumor invasion and metastatic potential, thereby degrading the extracellular matrix and allowing cells to cross the basement membrane, facilitating tumor cell metastasis [31][32]. Blocking ERK/MAPK activation inhibited cell migration and stem characteristics of the nhri-hn1 cell line in a mouse tongue cancer model [33]. Junhai et al. found that miR-145 could inactivate the ERK/MAPK signaling pathway by inhibiting Hoxa1, thereby inhibiting the proliferation, migration, and invasion of OSCC cells and inhibiting their growth in vivo [34]. The integrin (ITG) family of proteins plays important roles in OSCC αV invasion, migration, and apoptosis via ITG-β. They regulate the proliferation and invasion of OSCC cells through the MAPK/ERK signaling pathway [35][36][37]. Chloride intracellular channel 1 (CLIC1) silence reduces αv and β1. p-ERK, vimentin, MMP2, and MMP9 levels increased p-p38, E-cadherin, Caspase3, and caspase9 levels. CLIC1 interacted with ITG, thereby activating the MAPK signaling pathway, which regulates OSCC progression [38]. Different MAPK activation times may lead to different results, occasionally even contradictory ones. For example, transient activation of ERK may generate proliferative signals, but sustained phosphorylation may generate signals leading to cell differentiation [39].
3.2. JNK/MAPK Signaling Pathway in OSCC
JNKs were isolated and identified as stress-activated protein kinases, which activate the inhibitory response to protein synthesis [40]. The JNK protein is encoded by three genes, i.e., MAPK8 (JNK1), MAPK9 (JNK2), and MAPK10 (JNK3), and alternately spliced to produce ≥ 10 isomers. JNK1 and JNK2 are expressed in almost every cell, while JNK3 is mainly expressed in the brain [41]. The carcinogenic function of JNKs is related to their ability to phosphorylate Jun and activate AP1. In contrast, their antitumor effect may be related to the apoptotic activity [42]. In addition, JNK1 and JNK2 play different roles in cancer, promoting or inhibiting tumor formation [43].
Although some studies support the carcinogenic effect of JNK, others show that JNK plays a tumor-inhibitory role in HNSCC [44], which requires more studies to clarify its role in OSCC.
3.3. p38/MAPK Signaling Pathway in OSCC
P38 kinase was originally screened and defined in drugs that inhibit the TNF-mediated inflammatory response [45]. The p38-MAPK pathway and some physiological changes in cells, such as growth signal transmission, the ability of unlimited replication, and apoptosis, angiogenesis, invasion, or metastasis prevention, are involved in transformation [46]. P38 is a conserved serine–threonine protein kinase, which can be activated by various extracellular inflammatory factors (e.g., TNF-α, IL-1), bacterial lipopolysaccharide, lipopolysaccharide, chemokine, and ultraviolet light. Activated p38 MAPK regulates cell function by regulating expression activities of downstream enzymes and transcription factors [47]. p38 MAPK activation is necessary for normal immune processes and inflammatory responses. It promotes key regulators of pro-inflammatory cytokine biosynthesis through transcription and translation, and thus, the components of this pathway become potential therapeutic targets for autoimmune and inflammatory diseases [48]. Simultaneously, it participates in tumorigenesis and ischemia-reperfusion injury [47]. In OSCC, p38 signal inhibition can reduce the tumor proliferation rate and reduce inflammation caused by the tumor [49][50]. Angiogenesis plays a key role in tumor progression, providing nutrition and oxygen for tumors and eliminating metabolic waste and carbon dioxide. Continuous neovascularization promotes tumor growth and diffusion [46][51]. P38α can control the growth of cancer cells and tumor-induced angiogenesis and lymphangiogenesis. It is a positive regulator in the tumor microenvironment of OSCC [12]. Banerjee et al. found that glycophorin receptor 2 (GALR2) induces angiogenesis by secreting pro-angiogenic cytokines mediated by the p38/MAPK signaling pathway, vascular endothelial growth factor (VEGF), and IL-6. In addition, GALR2 activates the small GTP protein Rap1b, which induces the inactivation of p38-mediated tristetraprolin (TTP). TTP is an RNA-BP that downregulates angiogenic factors, such as IL-6, VEGF, and IL-8, produced by tumor and inflammatory cells [52][53][54]. Its function is to destroy the stable transcription of cytokines, increase the secretion of pro-angiogenic cytokines, and promote angiogenesis in vitro and in vivo. In OSCC cells with GALR2 overexpression, p38 inhibition activates TTP and reduces cytokine secretion. TTP inactivation increases IL-6 and VEGF secretions [13]. IL-6 is a biomarker with low disease-specific survival [55], and VEGF elevation is associated with reduced recurrence time [56]. In HNSCC cases with low differentiation, p38 activation is more obvious and associated with a poor prognosis. The P38/MAPK signaling pathway is related to apoptosis and autophagy. Treating cells with the p38 MAPK (SB203580) or JNK1/2 (sp600125) inhibitor can promote/weaken G2/M phase arrest, apoptosis, and autophagy of cancer cells, respectively [57].
3.4. MAPK Signaling Pathway and Immunity
Tumor cells downregulate immune cells in the tumor microenvironment to obtain tumor-promoting activity. MAPK is a central molecule of signal transduction regulating cell function. P38a MAPK participates in inflammation. It can produce proinflammatory cytokines [58], and acute inflammation can lead to cancer [59]. In the inflammatory microenvironment of OSCC, p38 αMAPK produces proinflammatory cytokines TNF-α, IL-1B, and IL-6, and plays a role in cancer progression [60]. Around MAPK-mutated HNSCC tumor cells, there exists a tumor microenvironment with high CD8+ T-cell inflammatory immunoreactivity, resulting in an increased endogenous lytic activity. These differences are evident in OSCC, suggesting that the ability of MAPK pathway mutation to predict disease in OSCC may be stronger than that of TMB. Pan pathway immune profiling studies revealed that MAPK-mutant tumors are the only “CD8+ T-cell inflammatory” tumors with an inherently hyperimmune responsive and structurally cytolytic tumor microenvironment. Immunoreactive MAPK-mutant models of HNSCC show massive in situ recruitment of cell-active or dead in situ CD8+ T cells. Consistent with the CD8+ T inflammatory phenotype, patients with MAPK-mutant OSCC had a 3.3–4.0-times longer survival time than patients with WT receiving anti-PD1/PD-L1 immunotherapy, independent of the tumor mutation burden. The pan-cancer prognosis of patients is consistent. MAPK mutations may recognize the high inflammatory/cytolytic activity of CD8+ T cells in patients with OSCC. p38 inhibitors have shown some success in treating and limiting adverse sequelae of inflammatory diseases and are a potential adjuvant therapy for OSCC [61].
IL-8 is closely related to the MAPK signaling pathway in OSCC. Leong et al. showed that it induces p-p38 MAPK and p-ERK expressions in HNSCC cells and downregulates p-JNK expression. It can increase NF-κB pathway expression in OSCC, suggesting that it may regulate MAPK and NF-κB pathways to regulate inflammatory response [11]. ROS or calcium ions activate p38MAPK, ERK [62], and JNK, which then affect the activation and transcriptional activity of hypoxia-inducible factor (HIF)-1. In addition, ERK leads to ser276p65/rela NF-κB phosphorylation of B, activating this transcription factor [63]. These processes lead to increased expression of some inflammatory genes and cell response to proinflammatory factors, particularly cyclooxygenase-2 [64], CC motif chemokine ligand 2/monocyte chemoattractant protein 1 (MCP-1) [65], CXC motif chemokine ligand (CXCL) 1/growth-related oncogene-α [66], CXCL8/IL-8 [67], and IL-6 [68]. They are inflammatory mediators involved in various tumor processes [69][70]. Figure 2 lists some well-studied key molecular pathways.
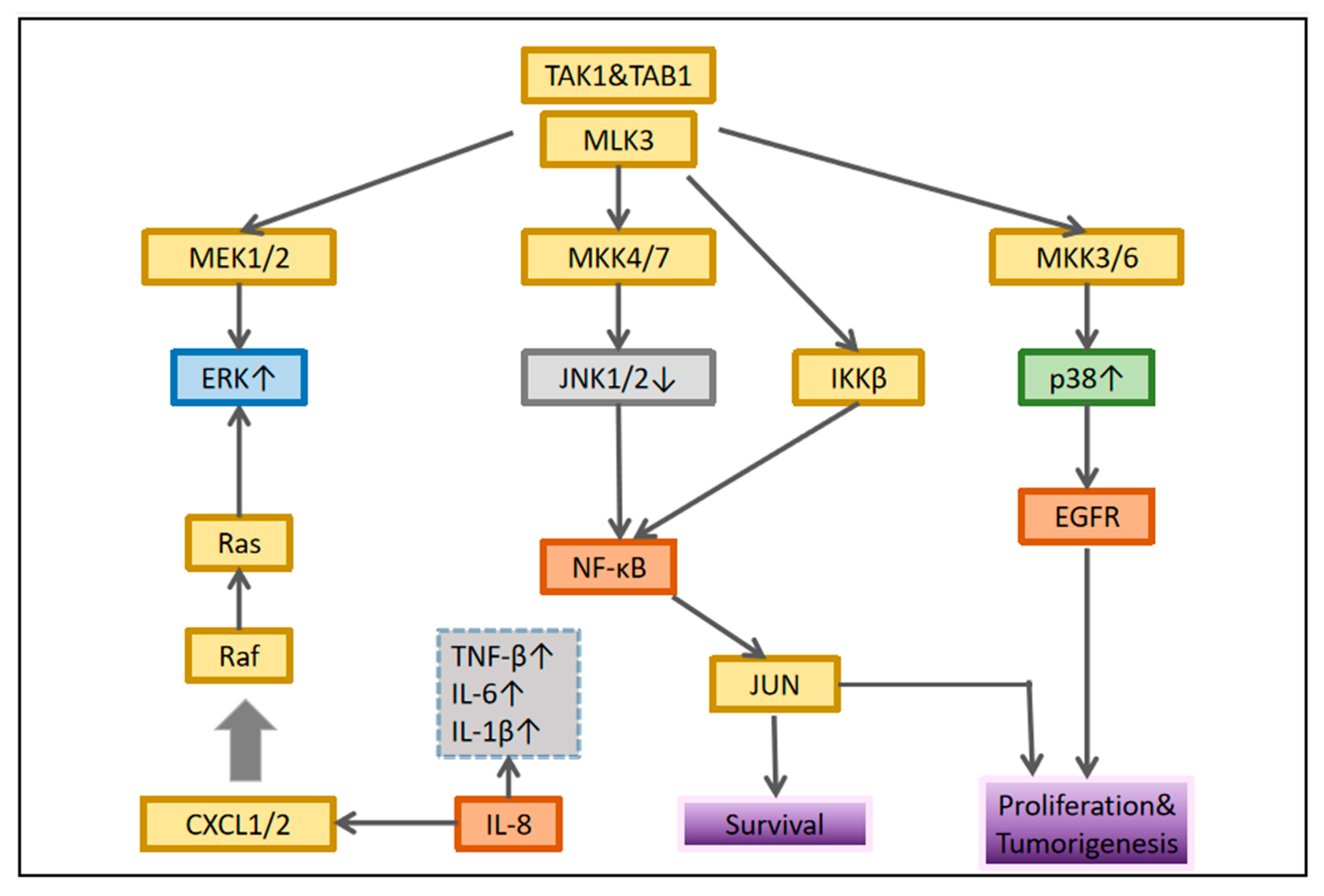
Figure 2. Interaction between MAPK and nuclear factor-κB signaling. In each cellular system, different connections are established that determine the biological response of the tumor.
4. MAPK Signaling Pathway in the EMT Process in OSCC
Overcoming intercellular adhesion and invading surrounding tissues is the main feature of the transformation from benign lesions to metastatic cancer. EMT is key to this transformation. It is molecularly characterized by loss of E-calmodulin and increased expression of mesenchymal markers, including, for example, n-calmodulin, snail, fibronectin, and wave proteins [71][72]. The loss of E-cadherin is closely related to a poor prognosis [73]. As a result, the discovery of potential EMT blockers in patients with OSCC may reveal directions for novel therapies. Snails can regulate transcriptional inhibition of epithelial marker E-cadherin during EMT [74]. MAPKP38 increased significantly in tumor cell lines with high snail expression. The P38 interacting protein (p38ip) is a human analogue of the yeast spt20 protein. It is a subunit of histone spt3-taf9-gcn5 acetyltransferase. P38 binds to and stabilizes p38ip, resulting in enhanced transcription. P38-p38ip is involved in snail-induced downregulation of E-cadherin and cell invasion in OSCC. The transcription inhibitor snail plays a direct role in the downregulation of E-cadherin, while the proinflammatory mediator upregulates snail, thus affecting the cycle of inflammation-promoting tumor progression [75]. Cui et al. [76]. found that protein kinase D3 regulates PD-L1 expression and EMT in OSCC through ERK1/2. In OSCC, transient knockout of ERK1/2 can cancel PD-1/PD-L1-induced EMT. Similarly, ERK1/2 activation is affected by PD-L1 knockdown. PD-L1 interacts with PD-1 expressed by activated T cells, B cells, natural killer cells, some dendritic cells, and tumor-associated macrophages, thereby activating the PD-1/PD-L1 pathway. In contrast, activation of this pathway can inhibit the anti-tumor function of the same immune cells, decreasing anti-tumor immunity [77][78].
This entry is adapted from the peer-reviewed paper 10.3390/cancers14194625
References
- Chang, L.; Karin, M. Mammalian MAP Kinase Signalling Cascades. Nature 2001, 410, 37–40.
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2020, 12, 491.
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK Signalling: A Master Regulator of Cell Behaviour, Life and Fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632.
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK Signalling Pathway and Tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007.
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857.
- Hirata, Y. Reactive Oxygen Species (ROS) Signaling: Regulatory Mechanisms and Pathophysiological Roles. Yakugaku Zasshi 2019, 139, 1235–1241.
- Imperial, R.; Toor, O.M.; Hussain, A.; Subramanian, J.; Masood, A. Comprehensive Pancancer Genomic Analysis Reveals (RTK)-RAS-RAF-MEK as A Key Dysregulated Pathway in Cancer: Its Clinical Implications. Semin. Cancer Biol. 2019, 54, 14–28.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404.
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1.
- Ngan, H.L.; Liu, Y.; Fong, A.Y.; Poon, P.H.Y.; Yeung, C.K.; Chan, S.S.M.; Lau, A.; Piao, W.; Li, H.; Tse, J.S.W.; et al. MAPK pathway mutations in head and neck cancer affect immune microenvironments and ErbB3 signaling. Life Sci. Alliance 2020, 3, e201900545.
- Chan, L.P.; Liu, C.; Chiang, F.Y.; Wang, L.F.; Lee, K.W.; Chen, W.T.; Kuo, P.L.; Liang, C.H. IL-8 Promotes Inflammatory Mediators and Stimulates Activation of p38 MAPK/ERK-NF-κB Pathway and Reduction of JNK in HNSCC. Oncotarget 2017, 8, 56375–56388.
- Leelahavanichkul, K.; Amornphimoltham, P.; Molinolo, A.A.; Basile, J.R.; Koontongkaew, S.; Gutkind, J.S. A Role for p38 MAPK in Head and Neck Cancer Cell Growth and Tumor-Induced Angiogenesis and Iymphangiogenesis. Mol. Oncol. 2014, 8, 105–118.
- Banerjee, R.; Van Tubergen, E.A.; Scanlon, C.S.; Vander Broek, R.; Lints, J.P.; Liu, M.; Russo, N.; Inglehart, R.C.; Wang, Y.; Polverini, P.J.; et al. The G Protein-Coupled Receptor GALR2 Promotes Angiogenesis in Head and Neck Cancer. Mol. Cancer Ther. 2014, 13, 1323–1333.
- Guo, Y.; Liu, C.; Zhang, J.; Tian, B.B.; Wang, L.; Liu, G.K.; Liu, Y. A Relationship between MAPK/ERK Pathway Expression and Neuronal Apoptosis in Rats with white Matter Lesions. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4412–4419.
- Uzgare, A.R.; Kaplan, P.J.; Greenberg, N.M. Differential Expression and/or activation of P38MAPK, erk1/2, and jnk during the Initiation and Progression of Prostate Cancer. Prostate 2003, 55, 128–139.
- Tsuji, T.; Miyazaki, M.; Sakaguchi, M.; Inoue, Y.; Namba, M. A REIC Gene Shows Down-Regulation in Human Immortalized Cells and Human Tumor-Derived Cell Lines. Biochem. Biophys. Res. Commun. 2000, 268, 20–24.
- Katase, N.; Nagano, K.; Fujita, S. DKK3 Expression and Function in Head and Neck Squamous Cell Carcinoma and Other Cancers. J. Oral. Biosci. 2020, 62, 9–15.
- Al Shareef, Z.; Kardooni, H.; Murillo-Garzón, V.; Domenici, G.; Stylianakis, E.; Steel, J.H.; Rabano, M.; Gorroño-Etxebarria, I.; Zabalza, I.; Vivanco, M.D.; et al. Protective Effect of Stromal Dickkopf-3 in Prostate Cancer: Opposing Roles for TGFBI and ECM-1. Oncogene 2018, 37, 5305–5324.
- Edamura, K.; Nasu, Y.; Takaishi, M.; Kobayashi, T.; Abarzua, F.; Sakaguchi, M.; Kashiwakura, Y.; Ebara, S.; Saika, T.; Watanabe, M.; et al. Adenovirus-Mediated REIC/Dkk-3 Gene Transfer Inhibits Tumor Growth and Metastasis in an Orthotopic Prostate Cancer Model. Cancer Gene Ther. 2007, 14, 765–772.
- Kawasaki, K.; Watanabe, M.; Sakaguchi, M.; Ogasawara, Y.; Ochiai, K.; Nasu, Y.; Doihara, H.; Kashiwakura, Y.; Huh, N.H.; Kumon, H.; et al. REIC/Dkk-3 Overexpression Downregulates P-Glycoprotein in Multidrug-Resistant MCF7/ADR Cells and Induces Apoptosis in Breast Cancer. Cancer Gene Ther. 2009, 16, 65–72.
- Katase, N.; Nishimatsu, S.I.; Yamauchi, A.; Yamamura, M.; Fujita, S. DKK3 Knockdown Confers Negative Effects on the Malignant Potency of Head and Neck Squamous Cell Carcinoma Cells via the PI3K/Akt and MAPK Signaling Pathways. Int. J. Oncol. 2019, 54, 1021–1032.
- Marconi, G.D.; Della Rocca, Y.; Fonticoli, L.; Melfi, F.; Rajan, T.S.; Carradori, S.; Pizzicannella, J.; Trubiani, O.; Diomede, F. C-Myc Expression in Oral Squamous Cell Carcinoma: Molecular Mechanisms in Cell Survival and Cancer Progression. Pharmaceuticals 2022, 15, 890.
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83.
- Aguzzi, A.; Maggioni, D.; Nicolini, G.; Tredici, G.; Gaini, R.M.; Garavello, W. MAP Kinase Modulation in Squamous Cell Carcinoma of the Oral Cavity. Anticancer Res. 2009, 29, 303–308.
- York, R.D.; Yao, H.; Dillon, T.; Ellig, C.L.; Eckert, S.P.; McCleskey, E.W.; Stork, P.J. Rap1 Mediates Sustained MAP Kinase Activation Induced by Nerve Growth Factor. Nature 1998, 392, 622–626.
- Rivera, C.; Zandonadi, F.S.; Sánchez-Romero, C.; Soares, C.D.; Granato, D.C.; González-Arriagada, W.A.; Paes Leme, A.F. Agrin has a Pathological Role in the Progression of Oral Cancer. Br. J. Cancer 2018, 118, 1628–1638.
- Chung, J.H.; Ostrowski, M.C.; Romigh, T.; Minaguchi, T.; Waite, K.A.; Eng, C. The ERK1/2 Pathway Modulates Nuclear PTEN-Mediated Cell Cycle Arrest by Cyclin D1 Transcriptional Regulation. Hum. Mol. Genet. 2006, 15, 2553–2559.
- Li, Y.; Xiang, Z.Y.; Xiong, J.; Hou, Z.W.; Zhu, Z.; Bao, W.W. RN181 Regulates the Biological Behaviors of Oral Squamous Cell Carcinoma Cells via Mediating ERK/MAPK Signaling Pathway. Acta Histochem. 2021, 123, 151733.
- Kang, H.M.; Park, B.S.; Kang, H.K.; Park, H.R.; Yu, S.B.; Kim, I.R. Delphinidin Induces Apoptosis and Inhibits Epithelial-to-Mesenchymal Transition via the ERK/p38 MAPK-Signaling Pathway in Human Osteosarcoma Cell Lines. Environ. Toxicol. 2018, 33, 640–649.
- Wu, H.; Li, L.; Ai, Z.; Yin, J.; Chen, L. Pristimerin Induces Apoptosis of Oral Squamous Cell Carcinoma Cells via G1 Phase Arrest and MAPK/Erk1/2 and Akt Signaling Inhibition. Oncol. Lett. 2019, 17, 3017–3025.
- Lin, C.W.; Chin, H.K.; Lee, S.L.; Chiu, C.F.; Chung, J.G.; Lin, Z.Y.; Wu, C.Y.; Liu, Y.C.; Hsiao, Y.T.; Feng, C.H.; et al. Ursolic Acid Induces Apoptosis and Autophagy in Oral Cancer Cells. Environ. Toxicol. 2019, 34, 983–991.
- Dong, Q.Z.; Wang, Y.; Tang, Z.P.; Fu, L.; Li, Q.C.; Wang, E.D.; Wang, E.H. Derlin-1 is Overexpressed in Non-Small Cell Lung Cancer and Promotes Cancer Cell Invasion via EGFR-ERK-Mediated Up-Regulation of MMP-2 and MMP-9. Am. J. Pathol. 2013, 182, 954–964.
- Chen, Y.L.; Liu, K.J.; Jang, C.W.; Hsu, C.C.; Yen, Y.C.; Liu, Y.L.; Chuang, T.H.; Wang, S.H.; Fu, Y.K.; Kuo, C.C.; et al. ERK Activation Modulates Cancer Stemness and Motility of a Novel Mouse Oral Squamous Cell Carcinoma Cell Line. Cancers 2019, 12, 61.
- Ding, J.; Sun, D.; Xie, P. Elevated microRNA-145 Inhibits the Development of Oral Squamous Cell Carcinoma through Inactivating ERK/MAPK Signaling Pathway by Down-Regulating HOXA1. Biosci. Rep. 2019, 39, BSR20182214.
- Veeravarmal, V.; Austin, R.D.; Nagini, S.; Nassar, M.H.M. Expression of β1integrin in Normal Epithelium, Oral Submucous Fibrosis and Oral Squamous Cell Carcinoma. Pathol. Res. Pract. 2018, 214, 273–280.
- Hayashido, Y.; Kitano, H.; Sakaue, T.; Fujii, T.; Suematsu, M.; Sakurai, S.; Okamoto, T. Overexpression of Integrin αv Facilitates Proliferation and Invasion of Oral Squamous Cell Carcinoma Cells via MEK/ERK Signaling Pathway that is Activated by Interaction of Integrin αvβ8 with Type I Collagen. Int. J. Oncol. 2014, 45, 1875–1882.
- Wang, S.H.; Liou, G.G.; Liu, S.H.; Chang, J.S.; Hsiao, J.R.; Yen, Y.C.; Chen, Y.L.; Wu, W.L.; Chang, J.Y.; Chen, Y.W. Laminin γ2-Enriched Extracellular Vesicles of Oral Squamous Cell Carcinoma Cells Enhance in Vitro Lymphangiogenesis via Integrin α3-Dependent Uptake by Lymphatic Endothelial Cells. Int. J. Cancer 2019, 144, 2795–2810.
- Feng, J.; Xu, J.; Xu, Y.; Xiong, J.; Xiao, T.; Jiang, C.; Li, X.; Wang, Q.; Li, J.; Li, Y. CLIC1 Promotes the Progression of Oral Squamous Cell Carcinoma via Integrins/ERK Pathways. Am. J. Transl. Res. 2019, 11, 557–571.
- Chambard, J.C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK Implication in Cell Cycle Regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310.
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The Stress-Activated Protein Kinase Subfamily of c-Jun Kinases. Nature 1994, 369, 156–160.
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Dérijard, B.; Davis, R.J. Selective Interaction of JNK Protein Kinase Isoforms with Transcription Factors. EMBO J. 1996, 15, 2760–2770.
- Weston, C.R.; Davis, R.J. The JNK Signal Transduction Pathway. Curr. Opin. Cell Biol. 2007, 19, 142–149.
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.; Xu, J.; Eriksson, S.; Hedström, E.; Issaeva, N.; Kel, A.; et al. ROS-Dependent Activation of JNK Converts p53 into an Efficient Inhibitor of Oncogenes Leading to Robust Apoptosis. Cell Death Differ. 2014, 21, 612–623.
- Boivin, A.; Hanot, M.; Malesys, C.; Maalouf, M.; Rousson, R.; Rodriguez-Lafrasse, C.; Ardail, D. Transient Alteration of Cellular Redox Buffering before Irradiation triggers Apoptosis in Head and Neck Carcinoma Stem and Non-Stem Cells. PLoS ONE 2011, 6, e14558.
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W.; et al. A Protein Kinase Involved in the Regulation of Inflammatory Cytokine Biosynthesis. Nature 1994, 372, 739–746.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Wagner, E.F.; Nebreda, A.R. Signal Integration by JNK and p38 MAPK Pathways in Cancer Development. Nat. Rev. Cancer 2009, 9, 537–549.
- Cuenda, A.; Rousseau, S. p38 MAP-Kinases Pathway Regulation, Function and Role in Human Diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375.
- Ozawa, H.; Ranaweera, R.S.; Izumchenko, E.; Makarev, E.; Zhavoronkov, A.; Fertig, E.J.; Howard, J.D.; Markovic, A.; Bedi, A.; Ravi, R.; et al. SMAD4 Loss Is Associated with Cetuximab Resistance and Induction of MAPK/JNK Activation in Head and Neck Cancer Cells. Clin. Cancer Res. 2017, 23, 5162–5175.
- Simon, C.; Goepfert, H.; Boyd, D. Inhibition of the p38 Mitogen-Activated Protein Kinase by SB 203580 Blocks PMA-Induced Mr 92,000 Type IV Collagenase Secretion and in vitro Invasion. Cancer Res. 1998, 58, 1135–1139.
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364.
- Van Tubergen, E.; Vander Broek, R.; Lee, J.; Wolf, G.; Carey, T.; Bradford, C.; Prince, M.; Kirkwood, K.L.; D’Silva, N.J. Tristetraprolin Regulates Interleukin-6, which is Correlated with Tumor Progression in Patients with Head and Neck Squamous Cell Carcinoma. Cancer 2011, 117, 2677–2689.
- Zhao, W.; Liu, M.; D’Silva, N.J.; Kirkwood, K.L. Tristetraprolin Regulates Interleukin-6 Expression through p38 MAPK-Dependent affinity Changes with mRNA 3’ Untranslated Region. J. Interferon Cytokine Res. 2011, 31, 629–637.
- Suswam, E.; Li, Y.; Zhang, X.; Gillespie, G.Y.; Li, X.; Shacka, J.J.; Lu, L.; Zheng, L.; King, P.H. Tristetraprolin Down-Regulates Interleukin-8 and Vascular Endothelial Growth Factor in Malignant Glioma Cells. Cancer Res. 2008, 68, 674–682.
- Márton, I.J.; Horváth, J.; Lábiscsák, P.; Márkus, B.; Dezső, B.; Szabó, A.; Tar, I.; Piffkó, J.; Jakus, P.; Barabás, J.; et al. Salivary IL-6 mRNA is a Robust Biomarker in Oral Squamous Cell Carcinoma. J. Clin. Med. 2019, 8, 1958.
- Eisma, R.J.; Spiro, J.D.; Kreutzer, D.L. Vascular Endothelial Growth Factor Expression in Head and Neck Squamous Cell Carcinoma. Am. J. Surg. 1997, 174, 513–517.
- Chen, J.C.; Hsieh, M.C.; Lin, S.H.; Lin, C.C.; His, Y.T.; Lo, Y.S.; Chuang, Y.C.; Hsieh, M.J.; Chen, M.K. Coronarin D Induces Reactive Oxygen Species-Mediated Cell Death in Human Nasopharyngeal Cancer Cells through Inhibition of p38 MAPK and Activation of JNK. Oncotarget 2017, 8, 108006–108019.
- Bradham, C.; McClay, D.R. p38 MAPK in Development and Cancer. Cell Cycle 2006, 5, 824–828.
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and Cancer: How Hot is the Link? Biochem. Pharmacol. 2006, 72, 1605–1621.
- Gill, K.; Kumar, R.; Mohanti, B.K.; Dey, S. Assessment of p38α in Peripheral Blood Mononuclear Cells (PBMC): A Potential Blood Protein Marker of Head and Neck Squamous Cell Carcinoma. Clin. Transl. Oncol. 2013, 15, 969–973.
- Medicherla, S.; Reddy, M.; Ying, J.; Navas, T.A.; Li, L.; Nguyen, A.N.; Kerr, I.; Hanjarappa, N.; Protter, A.A.; Higgins, L.S. p38alpha-Selective MAP Kinase Inhibitor Reduces Tumor Growth in Mouse Xenograft Models of Multiple Myeloma. Anticancer Res. 2008, 28, 3827–3833.
- Yadav, S.; Kalra, N.; Ganju, L.; Singh, M. Activator Protein-1 (AP-1): A Bridge between Life and Death in Lung Epithelial (A549) Cells under Hypoxia. Mol. Cell Biochem. 2017, 436, 99–110.
- Scortegagna, M.; Cataisson, C.; Martin, R.J.; Hicklin, D.J.; Schreiber, R.D.; Yuspa, S.H.; Arbeit, J.M. HIF-1alpha Regulates Epithelial Inflammation by Cell Autonomous NFkappaB Activation and Paracrine Stromal Remodeling. Blood 2008, 111, 3343–3354.
- Gutsche, K.; Randi, E.B.; Blank, V.; Fink, D.; Wenger, R.H.; Leo, C.; Scholz, C.C. Intermittent Hypoxia Confers Pro-Metastatic Gene Expression Selectively through NF-κB in Inflammatory Breast Cancer Cells. Free Radic. Biol. Med. 2016, 101, 129–142.
- Chuang, L.P.; Chen, N.H.; Lin, Y.; Ko, W.S.; Pang, J.H. Increased MCP-1 Gene Expression in Monocytes of Severe OSA Patients and under Intermittent Hypoxia. Sleep Breath 2016, 20, 425–433.
- Olbryt, M.; Habryka, A.; Student, S.; Jarząb, M.; Tyszkiewicz, T.; Lisowska, K.M. Global Gene Expression Profiling in Three Tumor Cell Lines Subjected to Experimental Cycling and Chronic Hypoxia. PLoS ONE 2014, 9, e105104.
- Song, D.; Fang, G.; Mao, S.Z.; Ye, X.; Liu, G.; Miller, E.J.; Greenberg, H.; Liu, S.F. Selective Inhibition of Endothelial NF-κB Signaling Attenuates Chronic Intermittent Hypoxia-Induced Atherosclerosis in Mice. Atherosclerosis 2018, 270, 68–75.
- Tellier, C.; Desmet, D.; Petit, L.; Finet, L.; Graux, C.; Raes, M.; Feron, O.; Michiels, C. Cycling Hypoxia Induces a Specific Amplified Inflammatory Phenotype in Endothelial Cells and Enhances Tumor-Promoting Inflammation in vivo. Neoplasia 2015, 17, 66–78.
- Korbecki, J.; Simińska, D.; Gąssowska-Dobrowolska, M.; Listos, J.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Chronic and Cycling Hypoxia: Drivers of Cancer Chronic Inflammation through HIF-1 and NF-κB Activation: A Review of the Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 10701.
- Ravenna, L.; Principessa, L.; Verdina, A.; Salvatori, L.; Russo, M.A.; Petrangeli, E. Distinct Phenotypes of Human Prostate Cancer Cells Associate with Different Adaptation to Hypoxia and Pro-Inflammatory Gene Expression. PLoS ONE 2014, 9, e96250.
- Na, T.Y.; Schecterson, L.; Mendonsa, A.M.; Gumbiner, B.M. The Functional Activity of E-Cadherin Controls Tumor Cell Metastasis at Multiple Steps. Proc. Natl. Acad. Sci. USA 2020, 117, 5931–5937.
- Aiello, N.M.; Kang, Y. Context-Dependent EMT Programs in Cancer Metastasis. J. Exp. Med. 2019, 216, 1016–1026.
- Zhou, B.; Xiang, J.; Jin, M.; Zheng, X.; Li, G.; Yan, S. High Vimentin Expression with E-Cadherin Expression Loss Predicts a Poor Prognosis after Resection of Grade 1 and 2 Pancreatic Neuroendocrine Tumors. BMC Cancer 2021, 21, 334.
- Tian, Y.; Qi, P.; Niu, Q.; Hu, X. Combined Snail and E-cadherin Predicts Overall Survival of Cervical Carcinoma Patients: Comparison Among Various Epithelial-Mesenchymal Transition Proteins. Front. Mol. Biosci. 2020, 7, 22.
- Lin, Y.; Mallen-St Clair, J.; Wang, G.; Luo, J.; Palma-Diaz, F.; Lai, C.; Elashoff, D.A.; Sharma, S.; Dubinett, S.M.; St John, M. p38 MAPK Mediates Epithelial-Mesenchymal Transition by Regulating p38IP and Snail in Head and Neck Squamous Cell Carcinoma. Oral. Oncol. 2016, 60, 81–89.
- Cui, B.; Chen, J.; Luo, M.; Liu, Y.; Chen, H.; Lü, D.; Wang, L.; Kang, Y.; Feng, Y.; Huang, L.; et al. PKD3 Promotes Metastasis and Growth of Oral Squamous Cell Carcinoma through Positive Feedback Regulation with PD-L1 and Activation of ERK-STAT1/3-EMT Signalling. Int. J. Oral. Sci. 2021, 13, 8.
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature 2017, 545, 495–499.
- Karyampudi, L.; Lamichhane, P.; Krempski, J.; Kalli, K.R.; Behrens, M.D.; Vargas, D.M.; Hartmann, L.C.; Janco, J.M.; Dong, H.; Hedin, K.E.; et al. PD-1 Blunts the Function of Ovarian Tumor-Infiltrating Dendritic Cells by Inactivating NF-κB. Cancer Res. 2016, 76, 239–250.
This entry is offline, you can click here to edit this entry!