Peptide mediated intracellular delivery of FADD protein, efficiently expressed in the cytosol and target core pro-tumorigenic NFκB signaling to restrict cancer cells proliferation. This approach has the potential to design strategies for targeted delivery of proteins inside the cells, which might be useful in cancer therapeutics.
- FADD therapy, Peptides, Cancer, NFkB, Drugs.
1. Introduction
The Fas-associated death domain (FADD) protein orchestrates several cellular pathways, including apoptosis, cell cycle regulation, autophagy, and inflammation[1][2]. The molecular structure of FADD consists of the N-terminal death effector domain (DED) and C-terminal death domain (DD). Upon activation of death receptors (DRs), the DD of FADD interacts with the DD of DRs, and facilitates the DED of FADD to oligomerize with DED containing pro-apoptotic proteins, such as procaspase-8/10, to form a multimeric death-inducing signaling complex (DISC)[3][4]. Indeed, the constituted DISC assembly concomitantly activates the canonical apoptosis signaling[5][6]. In cancer cells, the DED-containing protein cFLIPL (cellular FLICE inhibitory protein) competitively exclude caspase-8 binding to FADD at DISC and inhibits apoptosis signaling[7][8]. Importantly, post-translational modifications (PTMs) and nuclear localization of FADD have been reported in cancer cells, which further challenge the pro-apoptotic competency of FADD to instigate apoptosis signaling[9][10][11]. In this context, earlier studies demonstrated that low expression of FADD in cancer cells further exacerbates the severity of disease[12][13][14][15]. Moreover, somatic mutation in FADD[16] and elevated expression of cFLIP[17] has been attributed in the pathogenesis of colon carcinoma. We previously reported that induced expression of FADD regulates cFLIPL expression and favors procaspase-8 binding to DISC, independent of DR stimulation[18][19][20]. Targeting FADD as a therapeutic candidate would be a promising approach to reinstate apoptosis signaling in cancer cells.
It is well established that the tumor microenvironment is enriched with various pro-tumorigenic stimuli, such as tumor necrosis factor-α (TNF-α), inflammatory cytokines, and growth factors, which constitutively activates NF-κB signaling and cell proliferation[21][22][23]. Noteworthy, a constitutive NF-κB activation, leads to aberrant transcriptional priming of anti-apoptotic genes, such as Bcl-2, TRAF2, cFLIP, cIAPs, and RIP1 which further abrogates apoptosis signaling[22][24]. On the other side, pathogen recognition receptor (PRR)-mediated NF-κB activation induces transcriptional priming of proinflammatory genes to maintain the tumor microenvironment [25][26]. In this context, the NLRP3 inflammasome complex consisting of NLRP3 (nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing receptor 3), ASC (Apoptosis-associated speck-like protein containing a CARD), and pro-caspase-1 protein facilitates the proteolytic processing of NF-κB-induced proinflammatoryIL-1β to promote tumor growth[27][28][29]. We and others previously reported that transient expression of FADD abrogates NF-κB activation[18][30]. A recent report revealed that canonical NLRP3 inflammasome activation induces the secretion of soluble FADD protein in human monocytes and macrophages [31]; however, the exact molecular mechanism of FADD, in the regulation of NLRP3 inflammasome signaling is less explored in the context of apoptosis.
Given the importance of FADD in apoptosis and pro-tumorigenic NF-κB signaling, FADD protein may demonstrate a tremendous potential to mitigate cancer progression. In this context, few studies investigated the vector-based FADD gene therapy approach in regulation of tumor growth[32][33][34] and apoptosis in synoviocytes[35]; however, adenoviral or vector-based approaches have limited control over protein expression and host-derived factors. Other novel ways to directly deliver FADD protein in cancer cells are less explored. Recent advancement of direct protein delivery to cells provides a novel way to enrich poorly expressed proteins to the intracellular compartments[36][37][38]. The recent developments in therapeutic applications of small cell-penetrating peptides (CPPs), such as TAT (trans-acting activator of transcription) peptides, have been successfully validated to transport macromolecules, such as nucleic acids and proteins, across the cell membrane[39][40].
2.TAT-FADD Efficiently Induces Apoptosis Compared with Conventional Apoptosis Inducers
Given that TAT-FADD has the potential to induce both the caspase-8 and mitochondrial signaling of apoptosis, we next compared the apoptotic competency of TAT-FADD with conventional inducers of the death receptor and mitochondrial apoptosis. To this aim, we selected the death receptor ligands CD 95L and TNF-α; pro-apoptotic molecules, such as etoposide and HA14–1; and the protein translational inhibitor cycloheximide (CHX) (Figure 1A). The HCT 116 cells were treated with the mentioned molecules and TAT-FADD individually for 3–12 h. We found that within 3 h of treatment, TAT-FADD showed more disintegrated cellular morphology (shown with arrows) compared to the ligands or/and drugs, and remained at the maximum post 12 h of treatment (Figure 1B). Next, we measured cell viability and apoptotic death. We found that TAT-FADD death-inducing and pro-apoptotic activity was comparable with the death ligand CD 95L and pro-apoptotic molecule HA14-1, and post 12 h of incubation, TAT-FADD demonstrated maximum effect (Figure 1C). We measured the mitochondrial membrane potential (MMP) and found a significant alteration in MMP of TAT-FADD-treated cells similar to CD 95L and HA14-1, post 3–12 h of treatment (Figure 1D). Next, we monitored the activation of PARP and caspase-7 as a confirmation of apoptotic instigation. Interestingly, at 3 h, we found processing and activation of PARP and caspases-7 only in TAT-FADD-treated cells as compared to remaining molecules. Moreover, at 6 and 12 h of incubation, the activation of PAPR and caspases-7 was comparable in all molecules and the same as TAT-FADD (Figure 1E). Moreover, we found that TAT-FADD synergistically enhances the pro-apoptotic effect when treated in combination with the death ligands CD95L or TNF-α in HCT116 cells. Together, these results suggest that TAT-FADD rapidly induces apoptosis signaling and shows a similar pro-apoptotic response as observed with conventional death-inducing ligands and molecules.
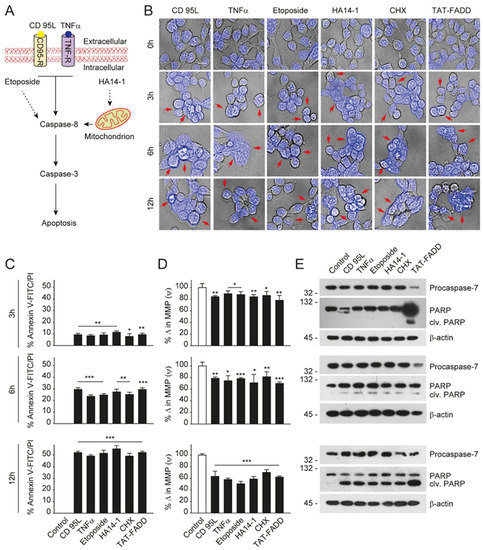
Figure 1. TAT-FADD’s pro-apoptotic effect is compared with conventional apoptosis inducers. (A) Schematic diagram representing the target site of proposed apoptosis inducers. (B–E) HCT 116 cells were treated with CD 95L (200 ng/mL), TNF-α (50 ng/mL), etoposide (50 µM), HA14-1 (5 µM), protein translational inhibitor cycloheximide (CHX, 5 µg/mL), and TAT-FADD (5 µM) alone for the mentioned time points, (B) The bright field images of cells counterstained with DAPI, post treatments, representative of 150 cells from 3 independent fields, scale bar 5 µm, (C) % apoptotic death by a Tali™ image-based cytometer, (D) % change in MMP and (E) expression of Procaspase-7 and cleavage of PARP by Western blot analysis, molecular weight marker left to each blot. In (C,D), significance compared between non-treated (0 h, white bars) and treated cells (black bars). h, hours; clv, cleaved; CD95-R, CD95 receptor; TNF-R, TNF receptor; MMP, mitochondrial membrane potential. Mean ± SD; * p ≤ 0.05, ** p ≤ 0.01, and *** p ≤ 0.001.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21186890
References
- Mouasni, S.; Tourneur, L. Fadd at the crossroads between cancer and inflammation. Trends Immunol. 2018, 39, 1036–1053.
- Werner, M.H.; Wu, C.; Walsh, C.M. Emerging roles for the death adaptor fadd in death receptor avidity and cell cycle regulation. Cell Cycle 2006, 5, 2332–2338.
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.J.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The fas-fadd death domain complex structure unravels signalling by receptor clustering. Nature 2009, 457, 1019–1022.
- Lavrik, I.; Krueger, A.; Schmitz, I.; Baumann, S.; Weyd, H.; Krammer, P.H.; Kirchhoff, S. The active caspase-8 heterotetramer is formed at the cd95 disc. Cell Death Differ. 2003, 10, 144–145.
- Ho, P.K.; Hawkins, C.J. Mammalian initiator apoptotic caspases. FEBS J. 2005, 272, 5436–5453.
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389.
- Okano, H.; Shiraki, K.; Inoue, H.; Kawakita, T.; Yamanaka, T.; Deguchi, M.; Sugimoto, K.; Sakai, T.; Ohmori, S.; Fujikawa, K.; et al. Cellular flice/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab. Investig. 2003, 83, 1033–1043.
- Ram, D.R.; Ilyukha, V.; Volkova, T.; Buzdin, A.; Tai, A.; Smirnova, I.; Poltorak, A. Balance between short and long isoforms of cflip regulates fas-mediated apoptosis in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1606–1611.
- Alappat, E.C.; Feig, C.; Boyerinas, B.; Volkland, J.; Samuels, M.; Murmann, A.E.; Thorburn, A.; Kidd, V.J.; Slaughter, C.A.; Osborn, S.L.; et al. Phosphorylation of fadd at serine 194 by ckialpha regulates its nonapoptotic activities. Mol. Cell 2005, 19, 321–332.
- Gomez-Angelats, M.; Cidlowski, J.A. Molecular evidence for the nuclear localization of fadd. Cell Death Differ. 2003, 10, 791–797.
- Lee, E.W.; Kim, J.H.; Ahn, Y.H.; Seo, J.; Ko, A.; Jeong, M.; Kim, S.J.; Ro, J.Y.; Park, K.M.; Lee, H.W.; et al. Ubiquitination and degradation of the fadd adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat. Commun. 2012, 3, 978.
- Chen, G.; Bhojani, M.S.; Heaford, A.C.; Chang, D.C.; Laxman, B.; Thomas, D.G.; Griffin, L.B.; Yu, J.; Coppola, J.M.; Giordano, T.J.; et al. Phosphorylated fadd induces nf-kappab, perturbs cell cycle, and is associated with poor outcome in lung adenocarcinomas. Proc. Natl. Acad. Sci. USA 2005, 102, 12507–12512.
- Marin-Rubio, J.L.; Vela-Martin, L.; Fernandez-Piqueras, J.; Villa-Morales, M. Fadd in cancer: Mechanisms of altered expression and function, and clinical implications. Cancers 2019, 11, 1462.
- Tourneur, L.; Delluc, S.; Levy, V.; Valensi, F.; Radford-Weiss, I.; Legrand, O.; Vargaftig, J.; Boix, C.; Macintyre, E.A.; Varet, B.; et al. Absence or low expression of fas-associated protein with death domain in acute myeloid leukemia cells predicts resistance to chemotherapy and poor outcome. Cancer Res. 2004, 64, 8101–8108.
- Tourneur, L.; Mistou, S.; Michiels, F.M.; Devauchelle, V.; Renia, L.; Feunteun, J.; Chiocchia, G. Loss of fadd protein expression results in a biased fas-signaling pathway and correlates with the development of tumoral status in thyroid follicular cells. Oncogene 2003, 22, 2795–2804.
- Soung, Y.H.; Lee, J.W.; Kim, S.Y.; Nam, S.W.; Park, W.S.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutation of fadd gene is rare in human colon and stomach cancers. APMIS 2004, 112, 595–597.
- Korkolopoulou, P.; Saetta, A.A.; Levidou, G.; Gigelou, F.; Lazaris, A.; Thymara, I.; Scliri, M.; Bousboukea, K.; Michalopoulos, N.V.; Apostolikas, N.; et al. C-flip expression in colorectal carcinomas: Association with fas/fasl expression and prognostic implications. Histopathology 2007, 51, 150–156
- Ranjan, K.; Pathak, C. Fadd regulates nf-kappab activation and promotes ubiquitination of cflipl to induce apoptosis. Sci. Rep. 2016, 6, 22787.
- Ranjan, K.; Surolia, A.; Pathak, C. Apoptotic potential of fas-associated death domain on regulation of cell death regulatory protein cflip and death receptor mediated apoptosis in hek 293t cells. J. Cell Commun. Signal. 2012, 6, 155–168.
- Ranjan, K.; Pathak, C. Expression of fadd and cflipl balances mitochondrial integrity and redox signaling to substantiate apoptotic cell death. Mol. Cell. Biochem. 2016, 422, 135–150.
- Li, Q.; Verma, I.M. Nf-kappab regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734.
- Xia, Y.; Shen, S.; Verma, I.M. Nf-kappab, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830.
- Eluard, B.; Thieblemont, C.; Baud, V. Nf-kappab in the new era of cancer therapy. Trends Cancer 2020, 6, 677–687.
- Nagel, D.; Vincendeau, M.; Eitelhuber, A.C.; Krappmann, D. Mechanisms and consequences of constitutive nf-kappab activation in b-cell lymphoid malignancies. Oncogene 2014, 33, 5655–5665.
- Sau, A.; Lau, R.; Cabrita, M.A.; Nolan, E.; Crooks, P.A.; Visvader, J.E.; Pratt, M.A. Persistent activation of nf-kappab in brca1-deficient mammary progenitors drives aberrant proliferation and accumulation of DNA damage. Cell Stem Cell 2016, 19, 52–65.
- Taniguchi, K.; Karin, M. Nf-kappab, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324.
- He, Y.; Hara, H.; Nunez, G. Mechanism and regulation of nlrp3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021.
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol. Cell 2002, 10, 417–426.
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832.
- Bannerman, D.D.; Tupper, J.C.; Kelly, J.D.; Winn, R.K.; Harlan, J.M. The fas-associated death domain protein suppresses activation of nf-kappa b by lps and il-1 beta. J. Clin. Investig. 2002, 109, 419–425.
- Mouasni, S.; Gonzalez, V.; Schmitt, A.; Bennana, E.; Guillonneau, F.; Mistou, S.; Avouac, J.; Ea, H.K.; Devauchelle, V.; Gottenberg, J.E.; et al. The classical nlrp3 inflammasome controls fadd unconventional secretion through microvesicle shedding. Cell Death Dis. 2019, 10, 190.
- Kondo, S.; Ishizaka, Y.; Okada, T.; Kondo, Y.; Hitomi, M.; Tanaka, Y.; Haqqi, T.; Barnett, G.H.; Barna, B.P. Fadd gene therapy for malignant gliomas in vitro and in vivo. Hum. Gene Ther. 1998, 9, 1599–1608.
- Komata, T.; Koga, S.; Hirohata, S.; Takakura, M.; Germano, I.M.; Inoue, M.; Kyo, S.; Kondo, S.; Kondo, Y. A novel treatment of human malignant gliomas in vitro and in vivo: Fadd gene transfer under the control of the human telomerase reverse transcriptase gene promoter. Int. J. Oncol. 2001, 19, 1015–1020.
- Yang, Y.W.; Zhang, C.M.; Huang, X.J.; Zhang, X.X.; Zhang, L.K.; Li, J.H.; Hua, Z.C. Tumor-targeted delivery of a c-terminally truncated fadd (n-fadd) significantly suppresses the b16f10 melanoma via enhancing apoptosis. Sci. Rep. 2016, 6, 34178.
- Kobayashi, T.; Okamoto, K.; Kobata, T.; Hasunuma, T.; Kato, T.; Hamada, H.; Nishioka, K. Novel gene therapy for rheumatoid arthritis by fadd gene transfer: Induction of apoptosis of rheumatoid synoviocytes but not chondrocytes. Gene Ther. 2000, 7, 527–533.
- Lee, Y.W.; Luther, D.C.; Kretzmann, J.A.; Burden, A.; Jeon, T.; Zhai, S.; Rotello, V.M. Protein delivery into the cell cytosol using non-viral nanocarriers. Theranostics 2019, 9, 3280–3292.
- Liu, X.; Wu, F.; Ji, Y.; Yin, L. Recent advances in anti-cancer protein/peptide delivery. Bioconjug. Chem. 2019, 30, 305–324.
- Zhang, Y.; Roise, J.J.; Lee, K.; Li, J.; Murthy, N. Recent developments in intracellular protein delivery. Curr. Opin. Biotechnol. 2018, 52, 25–31.
- Jafari, B.; Pourseif, M.M.; Barar, J.; Rafi, M.A.; Omidi, Y. Peptide-mediated drug delivery across the blood-brain barrier for targeting brain tumors. Expert Opin. Drug Deliv. 2019, 16, 583–605. [Google Scholar] [CrossRef]
- Jones, S.W.; Christison, R.; Bundell, K.; Voyce, C.J.; Brockbank, S.M.; Newham, P.; Lindsay, M.A. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br. J. Pharm. 2005, 145, 1093–1102.