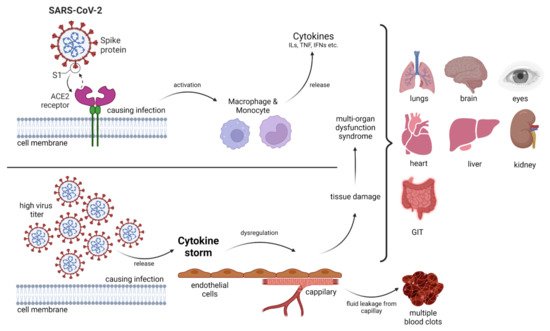
2. Structure of Coronavirus
SARS-CoV-2, a member of the β-coronavirus genus in the family Coronaviridae, has an envelope and positive-sense ssRNA genome of 29,891 nucleotides, encoding circular nucleocapsid proteins with 9860 amino acid residues [
21]. The viral particle size ranges from 80 to 220 nm. Overall, 10 open reading frames (ORFs) have been identified in its genome to date (approximately 26–32 kb). The first ORF (almost 2/3 of the viral RNA) encodes polyproteins 1a (ORF1a) and 1b (ORF1b) [
22]. Furthermore, these ORFs are cleaved by proteases into 16 nonstructural proteins (NSPs) that are responsible for genome replication and transcription [
23]. Structural proteins (SPs) are encoded by the remaining ORFs [
24,
25]. The main SPs and NSPs of SARS-CoV-2 are summarized in
Table 1 and
Table 2, respectively. The name coronavirus is derived from the appearance under the electron microscope, in which the presence of crown-like spikes on the envelope resembles the corona of the sun [
26]. SPs form the viral envelope that holds the RNA genome, while NSPs are expressed in host-infected cells but are not incorporated into virion infectious particles. These NSPs include various transcription factors and enzymes such as RNA-dependent RNA polymerase (RdRp) and hemagglutinin esterase (HE). Moreover, the virion employs enzymes such as RNA replicases and viral proteases to replicate itself [
22,
27,
28,
29].
Various SPs have been identified including the glycoprotein membrane (M), spike (S), small envelope (E), and nucleoprotein (N), and other accessory proteins. M-glycoprotein is the most abundant, spanning the membrane bilayer thrice [
30]. S-glycoprotein (150 kDa) is a type-I TM protein on the outer surface of the virus and is responsible for the binding of the virus to host cell receptors (ACE2). The S protein amino acid sequence of SARS-CoV-2 exhibits 86% similarity to that of SARS-CoV [
31]. The S protein consists of oligosaccharides bound to serine amino acids through
o-glycosides. The three major segments of S protein are the ectodomain, TM, and intracellular regions. The intracellular domain comprises the membrane fusion subunit S2 (trimeric stalk) as well as a short tail part known as the receptor-binding S1 domain (RBD; three S1 heads) [
32,
33]. Protein–protein interaction (PPI) between the human ACE2 and SARS-CoV-2 S protein facilitates viral attachment as well as the cellular entry of coronaviruses; thus, small-molecule blockage of these PPIs is a more inspiring therapeutic approach than inhibition via antibodies [
34]. The S1 subunit of the S protein enables ACE2-mediated virus attachment, whereas the S2 subunit facilitates membrane fusion. Specifically, asparagine, glutamine, serine, phenylalanine, and leucine residues present in the S protein boost ACE2 binding [
35].
Moreover, N protein bound to nucleic acids is an important structural component of the virus, which is responsible for viral replication and cellular response to infection in the host cellular machinery [
31] (
Table 1). The N protein comprises a serine-rich linker region sandwiched between the N-terminal domain (NTD) and the C-terminal domain (CTD). These termini are crucial for viral entry and processing in host cells. The CTD regulates nucleocapsid formation and the NTD adheres to the viral genome in the form of orthorhombic crystals. Phosphorylation sites are also present in the linker region, which control its function [
35]. In the case of SARS-CoV, the N protein enhances the activation of cyclooxygenase-2 (COX-2), resulting in the inflammation of pulmonary cells [
36]. Moreover, the N protein interacts with the p42 proteasome subunit, which degrades the virion [
37]. This also disables type-I IFN, which is responsible for suppressing the host immune responses produced by biological systems against viral infections [
38]. The interaction of the N protein with heterogeneous nuclear ribonucleoproteins leads to increased viral RNA synthesis [
39]. The N protein sequence of SARS-CoV-2 shows a 94.3% similarity to that of the SARS-CoV [
31].
The smallest TM structural protein in coronaviruses is the E protein (
Table 1), which comprises two different domains: the NTD (1–9 residues) as well as a hydrophobic domain (10–37 residues), with a chain at the terminal (38–76 residues) [
40,
41,
42]. The E protein plays a crucial biological role, not only in the structural integrity of the virus, but also in host virulence [
43]. The E protein sequence of SARS-CoV-2 shows a 96.1% similarity to that of SARS-CoV [
31].
The M protein plays a crucial role in maintaining the shape of the viral envelope (
Table 1). This function can be achieved by interacting with other viral proteins that exhibit PPIs [
44]. The M protein is also known as the central organization of coronavirus proteins. The binding of E to M produces the virus envelope, and this interaction is sufficient for the synthesis and release of viruses [
45,
46]. The binding of M with S is an important event for the retention of the S protein in the endoplasmic reticulum–Golgi complex as well as its integration into new viruses [
46,
47]. Moreover, the interaction of N with M stabilizes the nucleocapsid (RNA–N protein complex) and the internal core of viruses, resulting in the completion of viral assembly [
47,
48]. The M protein amino acid sequence of SARS-CoV-2 exhibits a 96.4% similarity with that of SARS-CoV [
31].
Table 1. The structural proteins (SPs) of coronaviruses and their physiological significance.
Table 2. The non-structural proteins (NSPs) of coronaviruses and their physiological significance.
3. Overview of TLR Signaling
Invading pathogens stimulate the release of proinflammatory mediators in response to infection (
Figure 1 and
Figure 2). Signaling networks are necessary for the protection of the host against invading microorganisms. TLR signaling dysregulation plays a central role in the development and progression of infection. Inflammatory secretory molecules including chemokines, ILs, IFNs, and tumor necrosis factor-alpha (TNF-α) are part and parcel of TLR signaling, resulting in the modulation of cellular characteristics such as apoptosis, immune response, and proliferation [
88,
89,
90]. Mitogen-activated protein kinases (MAPKs) and NF-κB are activated by TLRs. TLR3 and TLR4 are involved in the stimulation of IRF3. In contrast, IRF7 is triggered by TLR7–9 [
91]. TLRs are stimulated by interactions with ligands to initiate an intracellular downstream signaling cascade, leading to activation of the host defense system [
92].
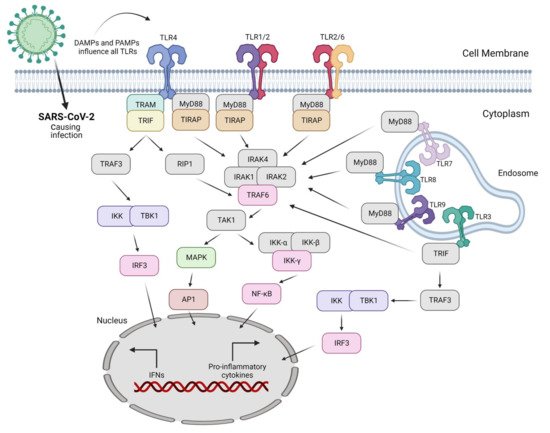
Figure 2. SARS-CoV-2 causes infection in the lungs mainly via DAMPs and PAMPs produced as a result of the action of nearly all Toll-like receptors (TLRs). Only TLRs involved in virus sensing and/or signaling are displayed here.
The nature of the ligand and downstream adaptor molecules directs the TLR signaling cascade (
Table 3). Two distinct pathways play critical roles in TLR signaling: MyD88-dependent and -independent pathways [
93] (
Figure 2). The former pathway employs all TLRs (except for TLR3), resulting in the biosynthesis of inflammatory cytokines [
94]. In contrast, the latter pathway (also referred to as the TRIF-dependent pathway) involves TLR3 and TLR4, resulting in the expression of IFN-I [
95]. In other words, the interaction of PAMP and PRR leads to the biosynthesis of proinflammatory cytokines as well as IFN-1, which is a cellular indication of the immune response [
96]. Several negative regulators that enhance the activation of the innate immune response are involved in TLR-dependent signaling cascades. Hence, the overactivation of TLRs can lead to the interruption of immune cell homeostasis, resulting in the risk of inflammatory disorders [
97]. Consequently, inhibitors (antagonists) targeting these receptors and/or cascades can serve as novel therapeutics to treat such disorders [
98].
Table 3. Toll-like receptors (TLRs) and their physiological significance.
4. Role of Antiviral Drugs Employing TLRs
When a pathogen such as a virus invades, an antiviral immune response is evident in the host cells. Various conserved molecular patterns of PAMPs have been identified. As discussed above, TLRs are the key constituents of the innate immune system, and multiple TLRs (TLR1–4, TLR6–9) identify viral ligands [
17,
117,
118,
119]. With respect to their functional importance, TLRs might be potentially employed to treat not only inflammatory disorders but also viral diseases. This can be explained by a deep insight into the positive and negative mediators of TLRs [
97,
120]. TLR agonists lack accessory molecules but can mimic natural ligands; hence, they exhibit a low molecular weight and have potential for expanded pharmacokinetics and pharmacodynamics in comparison with the parent molecule. Moreover, TLR antagonists help to deal with autoimmune and inflammatory disorders by defeating unnecessary inflammation, resulting in an antibody- or cell-mediated response that suppresses disease progression [
97,
121,
122].
Different approaches are employed by viruses in which they weaken their recognition by masking and/or increasing the dysregulation of mediators. Viruses disturb TLR signaling through their own mechanisms. Thus, TLRs are largely involved in the molecular interaction between viruses and host cells [
5]. Various PRRs are engaged in the response to viral infection, which is also the case for TLRs. A thorough understanding of this interaction has facilitated the development of various strategies to limit viral infection including antiviral immunity as well as therapeutics [
5]. Moreover, viral infection activates TLRs to increase cytokine levels, resulting in an antiviral innate immune response. The interaction between viruses and TLRs at every step of the signaling pathway plays an important role in developing effective antiviral therapies as well as in identifying novel molecular targets for the advancement in antiviral drugs [
123]. The regulation of invasion, replication, and immune responses is a significant factor in viral pathogenesis [
117]. Viral glycoproteins and NSPs released in the extracellular region are responsible for the stimulation of TLR2 and TLR4 due to their presence on the cellular surface [
117,
124,
125]. In contrast, TLR3, TLR7/8, and TLR9, which are present in the endosomal compartment, contain viral double-stranded RNA (dsRNA) [
126], ssRNA [
114], and CpG DNA (unmethylated) [
116], respectively.
TLR agonists have a positive effect on antiviral immunity and exhibit significant resistance against experimental infections [
127,
128,
129]. The TLR–virus interaction involves a complex mechanism that is associated with the type of TLR as well as the type of virus. Moreover, multiple PRRs are required to initiate an immune response to various viral infections. Moreover, significant differences in TLR signaling have been reported between mice and humans. Therefore, therapeutic manipulation of TLRs requires an understanding of human cellular immunity [
130]. Some examples are presented below.
TLR2 activation enhances the innate immune response to viral infections and can be used to treat viral respiratory diseases. Using the shock-and-kill strategy, immune cell recognition is enhanced and latently infected cells are eliminated [
112,
131]. TLRs can be used to reverse HIV-1 latency and trigger innate immune responses. In an evaluation of the effectiveness of SMU-Z1 (a novel TLR1/2 agonist), in addition to enhancing latent HIV-1 transcription (ex vivo), the NF-κB and MAPK pathways were also targeted in cells [
131]. Latency-reversing agents have been employed for HIV reactivation, resulting in enhanced immune activation [
112]. Dual TLR2/7 agonists were synthesized and characterized based on their latency-reversing ability, which were found to effectively reactivate the latency. TLR2 components reactivate HIV by NF-κB stimulation and the secretion of IL-22 (thereby enhancing the antiviral state and inhibiting HIV infection), whereas TLR7 components induce the secretion of TNF-α [
112]. The activation of TLR2 in vivo has been assessed against rhinovirus infection [
132]. Airway epithelial cells promote an extended immune response characterized by IFN-λ expression, NF-κB activation, and lymphocyte recruitment, resulting in a reduction in viral-induced inflammation and continued antiviral innate immunity [
132].
TLR3 (the first identified antiviral TLR) in humans confers protective immunity against vaccinia virus (VACV) infection. In contrast, TLR3 is responsible for the detrimental effects of VACV infection in mice and TLR4 has the same effect in humans [
133,
134]. The recognition of dsRNA by TLR3 is further evidence of the role of TLRs in the antiviral response [
119,
126,
135]. TLR3 signaling can be activated by a synthetic dsRNA agonist (a potent immune stimulant), resulting in protective immunity against multiple viruses including coronaviruses [
136,
137,
138,
139]. Viral-origin ssRNA sequences (rich in GU- and AU-) are detected by TLR7 and TLR8, which are functionally similar and only differ with respect to their expression patterns [
113,
130]. TLR7/8 expression is evident in dendritic cells, monocytes, and macrophages [
140]. Additional examples are listed in
Table 4.
Table 4. Reported antiviral agonists employing Toll-like receptors (TLRs).
|
Drugs
|
TLRs
|
Viruses
|
Significance
|
References
|
|
Pam2CSK4
|
TLR2
|
Parainfluenza
|
Reduced virus replication
|
[141]
|
|
INNA-051
|
TLR2
|
SARS-CoV-2
|
Reduces viral RNA load
|
[142]
|
|
PIKA
|
TLR3
|
Influenza A
|
Reduces virus load
|
[143]
|
|
Poly ICLC
|
TLR3
|
HIV
|
Release of IFN-α/β/γ
|
[144]
|
|
NA6
|
TLR4
|
Norovirus
|
Induction of IFN-β
|
[145]
|
|
MPL
|
TLR4
|
VZV
|
Stimulate cytokines
|
[146]
|
|
Flagellin
|
TLR5
|
Influenza A
|
Reduces virus replication
|
[147]
|
|
CBLB502
|
TLR5
|
ConA
|
Activation of NF-κB
|
[148]
|
|
Pam2CSK4
|
TLR6
|
Parainfluenza
|
Reduces virus replication
|
[141]
|
|
INNA-051
|
TLR6
|
SARS-CoV-2
|
Reduces viral RNA load
|
[142]
|
|
GS-9620
|
TLR7
|
HIV
|
Reactivates latency
|
[112]
|
|
Vesatolimod
|
TLR7
|
HIV
|
Modest delay in viral rebound
|
[149]
|
|
R848
|
TLR7/8
|
Zika
|
Activation of NF-κB
|
[150]
|
|
GS-9688
|
TLR8
|
HBV
|
Activation of dendritic and natural killer cells
|
[151]
|
|
ODN2395
|
TLR9
|
Parainfluenza
|
Reduces viral replication
|
[141]
|
CBLB502—Entolimod; ConA—Concanavalin A; GS-9688—Selgantolimod; R848—Resiquimod; NA6—neoagarohexaose; VZV—Varicella-Zoster virus.