Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Indane-1,3-dione is a versatile building block used in numerous applications ranging from biosensing, bioactivity, bioimaging to electronics or photopolymerization. Indane-1-3-dione is among one of the most privileged scaffolds in chemistry, as the derivatives of this structure can find applications in various research fields ranging from medicinal chemistry, organic electronics, photopolymerization, to optical sensing and non-linear optical (NLO) applications.
- indanedione
- chemical modification
- domino reaction
- MCR
- spiro compounds
1. Introduction
Indane-1-3-dione is among one of the most privileged scaffolds in chemistry, as the derivatives of this structure can find applications in various research fields ranging from medicinal chemistry, organic electronics, photopolymerization, to optical sensing and non-linear optical (NLO) applications. One of its closest analogues, namely indanone, is commonly associated with the design of biologically active compounds [1][2][3]. The most relevant examples in this field are undoubtedly Donepezil, which is still under use for the treatment of Alzheimer’s disease [4], or Indinavir, which is used for the treatment of AIDs disease [5]. Interest for indanone derivatives is notably motivated by the fact that this structure can be found in numerous natural products (Caraphenol B isolated from Caragna sinica [6], Pterosin B isolated from marine cyanobacterium [7], another derivative extracted from filamentous marine cyanobacterium Lyngbya majuscula) [7], sustaining the interest for these compounds [8][9]. Due to the similarity of structure with indanone, indane-1,3-dione is also of high current interest, and this molecule has also been extensively studied as a synthetic intermediate for the design of many different biologically active molecules [10]. Beyond its common use in the design of biologically active molecules, indane-1,3-dione is also an electron acceptor widely used for the design of dyes for solar cells applications, photoinitiators of polymerization or chromophores for NLO applications [11]. As an interesting feature, indane-1,3-dione possesses an active methylene group, making this electron acceptor an excellent candidate for its association with electron donors by means of Knoevenagel reactions [12]. Ketone groups can also be easily functionalized with malononitrile, enabling to convert it as a stronger electron acceptor.
2. Chemical Modification of the Indane-1,3-Dione Core
2.1. Synthesis of Indane-1,3-Dione
Indane-1,3-dione can be synthesized following different synthetic procedures. Furthermore, the most straightforward one consists in the nucleophilic addition of alkyl acetate 2 on dialkyl phthalate 1 under basic conditions, enabling to produce the intermediate 2-(ethoxycarbonyl)-1,3-dioxo-2,3-dihydro-1H-inden-2-ide anion 3. Then, by heating under acidic conditions, this intermediate can be hydrolyzed and decarboxylated in situ, producing indane-1,3-dione 4 in ca. 50% yield for the two steps [13][14]. However, several procedures were also reported to access 4 by oxidation of indane 5, using various oxidizing systems such as N-hydroxyphthalimide (NHPI) and tert-butyl nitrite (t-BuONO) [15], H2O2 with a Mn catalyst [16], pyridinium dichromate (PCC) in the presence of Adogen 464 and sodium percarbonate (Na2CO3·1.5 H2O2) [17]. Furthermore, in these different cases, reaction yields remained often limited (17 and 18% yields) while requiring expensive reagents so that the first procedure remains undoubtedly the most popular one to obtain indane-1,3-dione 4 in acceptable yield (see Scheme 1). Recently, a two-step procedure was developed, starting from 2-ethynylbenzaldehyde 6 [18]. By means of a Cu-catalyzed intramolecular annulation reaction, 3-hydroxy-2,3-dihydro-1H-inden-1-one 7 was prepared in 87% yield and a subsequent oxidation of 7 with Jones’ reagent enabled to obtain 4 in 95% yield. As alternative, o-iodoxybenzoic acid (IBX) can also be used as an oxidant, providing 4 in similar yield (92%) [19]. Several strategies were also developed to convert phthalic anhydride 8 into 4 using diethyl malonate 9 and montmorillonite KSF clay [20] or ethyl acetoacetate 11 in the presence of acetic anhydride and triethylamine [21]. In the case of substituted indane-1,3-diones, two distinct routes were developed depending on the substituents attached on the phthalic anhydrides. Thus, in the case of electron-withdrawing groups such as nitro and chlorine (12–14), the condensation of malonic acid in pyridine proved to be a straightforward route to access 19–21 [22]. Conversely, in the case of electron-donating groups such as alkyl substituents (22, 23), a specific procedure was developed, consisting [23] in a Friedel–Craft reaction of 4-methylbenzoyl chloride 22 or 3,4-dimethylbenzoyl chloride 23 with malonyl dichloride 24, followed by an acidic treatment with concentrated hydrochloric acid furnishing after purification of the two compounds 25 and 26 in 33 and 15% yield, respectively (see Scheme 2).
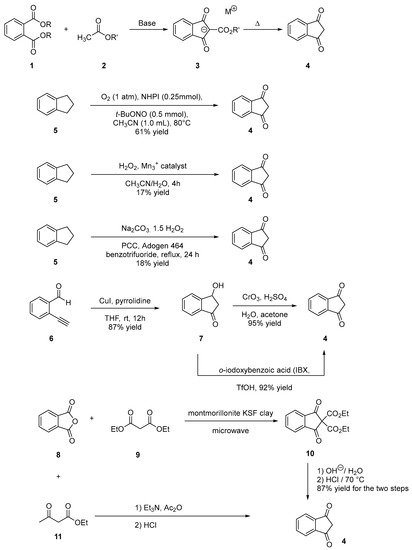
Scheme 1. Synthetic routes to indane-1,3-dione 4.
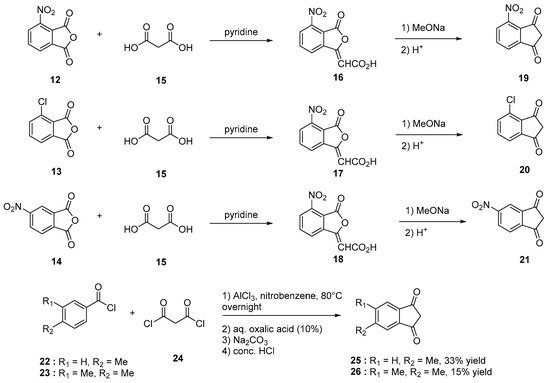
Scheme 2. Synthetic routes to substituted indane-1,3-diones 19–21 and 25, 26.
2.2. Chemical Engineering around the Ketone Groups
2.2.1. Functionalization with Cyano Groups
2-(3-Oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile 28 [24][25][26][27][28] and 2,2′-(1H-indene-1,3(2H)-diylidene)dimalononitrile 29 [28][29][30] can be synthesized by a Knoevenagel reaction of malononitrile 27 on 4 in ethanol using sodium acetate or piperidine as the bases. In the two cases, an excess of malononitrile was used, and the selection between the di- and the tetracyano-substituted derivative could be obtained by controlling the reaction temperature. Thus, the dicyano compound 28 can be obtained at room temperature contrarily to the tetracyano one 29 that is synthesized by heating the reaction media. In the case of 28, reaction yields ranging between 61 and 85% were determined, whereas 29 could be obtained with reaction yields ranging from 34 to 45% yield. Functionalization of substituted indane-1,3-diones (30, 32, 34) with only one dicyanomethylene group was also examined, and several situations were found. Thus, the Knoevenagel reaction furnished a mixture of inseparable isomers 31,31′, 33,33′ and 35,35′ when 5-methyl-1H-indene-1,3(2H)-dione 30 [23], ethyl 1,3-dioxo-2,3-dihydro-1H-indene-5-carboxylate 32 [31] or 5-fluoro-1H-indene-1,3(2H)-dione 34 [32] were used as the starting materials (see Scheme 3).
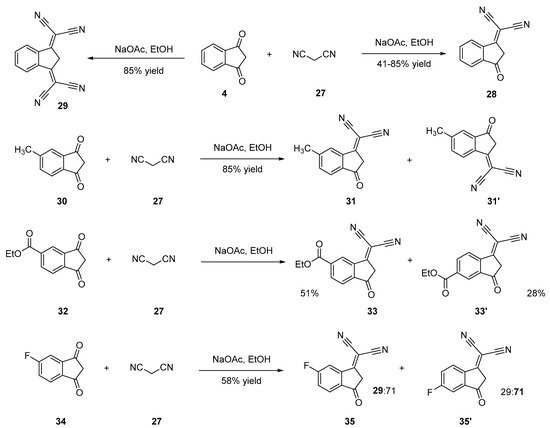
Scheme 3. Synthetic routes to 28, 31, 31′, 33, 33′, 35, 35′.
Conversely, 5-alkoxy-1H-indene-1,3(2H)-diones 36a furnished selectively 37a in 63% yield as a result of the specific activation of one of the two ketones by mesomeric effects [33]. A similar behavior was also observed during the synthesis of 39 [32], 41 [34] and 43 [35], their precursors 38, 40 and 42 being substituted with halogens. The steric hindrance generated by the substituents was another strategy to control the regioselectivity, and the synthesis of 45 is a relevant example of this (see Scheme 4) [36]. Concerning the symmetrically substituted indane-1,3-diones, those bearing halogens at the 5,6-positions were the most commonly studied, as exemplified with compounds 47 [37] or 49 [32][38].
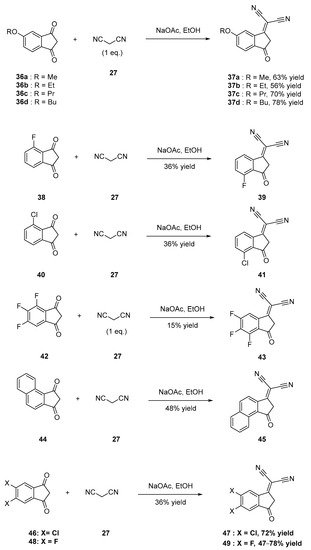
Scheme 4. Synthetic routes to 37a–37d, 39, 41, 43, 45, 47 and 49.
2.2.2. Self-Condensation of Indane-1,3-Dione: The Bindone Adduct
Bindone 50 is an electron acceptor widely used due to its stronger electron-withdrawing ability compared to that of 29. It can be easily synthesized by self-condensation of indane-1,3-dione 4 in basic conditions (triethylamine [25], sodium acetate [39], sodium hydride [40]), or in acidic conditions (sulfuric acid [41]) (see Scheme 5).
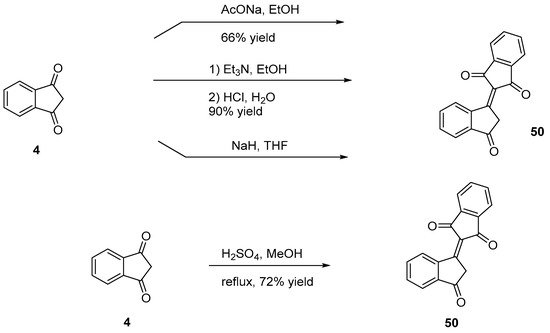
Scheme 5. Synthesis of bindone 50.
2.2.3. Formation of bis-Thiazoles and bis-Thiazolidinone
Indane-1,3-dione 4 and corresponding derivatives have been extensively studied for their biological activities ranging from antitumor, antibacterian and anti-inflammatory activities [13][42][43][44][45]. Parallel to this, 1,3,4-thiadiazole groups also exhibit biological activities [46][47][48][49][50][51][52][53][54] so that their combinations were examined [55]. From a synthetic viewpoint, bis-thiazoles could be obtained in two steps starting from indane-1,3-dione 4. By first reacting 4 with hydrazinecarboxamide 51 in ethanol, in the presence of triethylamine, 2,2′-((1H-indene-1,3(2H)-diylidene)bis(hydrazine-1-carboxamide) 52 could be obtained. Then, upon reaction with a number of N-aryl-2-oxopropane-hydrazonoyl chloride derivatives 53–55, bis-thiazoles 56–58 could be isolated with reaction yields ranging from 78 to 89%. Using a similar procedure, reaction of 52 with ethyl (N-arylhydrazono)chloroacetate 59–61 could furnish the corresponding bis-thiazolidinone 62–64 in high yields (79–90%) (see Scheme 6).
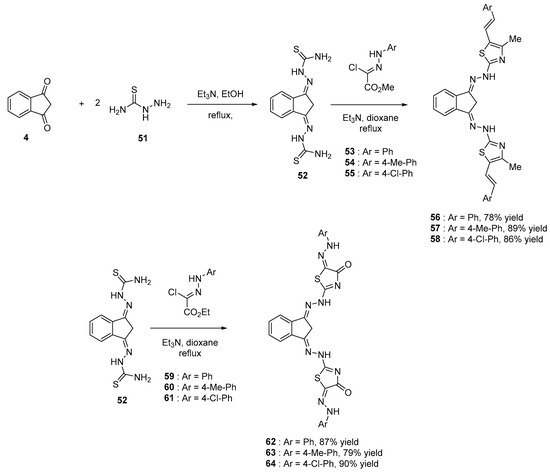
Scheme 6. Synthetic route to bis-thiazoles and bis-thiazolidinone starting from 4.
2.3. Chemical Engineering around the Aromatic Groups
2.3.1. Polyaromatic Structures
To improve the electron-accepting ability of 4, an alternative to the substitution of indane-1,3-dione with malononitrile consists in developing polyaromatic structures. Notably, naphthalene derivatives were prepared, following the same synthetic route to that used for 4, consisting in the condensation of ethyl acetate 66 on diethyl naphthalene-2,3-dicarboxylate 65 in solvent-free and basic conditions. After decarboxylation, 67 could be prepared in 91% yield for the two steps [12][56][57][58][59][60]. Introduction of lateral groups onto 67 is possible but involves a specific route to be developed. A Diels–Alder reaction between l,3-diphenylbenzo[c]furan 69 and cyclopent-4-ene-l,3-dione 70 furnishes the 1,3-diphenylbenzo[c]furan-cyclopent-4 ene-1,3-dione adduct 71. By dehydration in acidic conditions (HCl/H2SO4), 71 could be converted to 72 in 25% yield. Recently, the design of a helical-shaped structure 75 was reported [36]. If the structure is innovative, the synthesis is identical to that used for 4, starting from dimethyl naphthalene-1,2-dicarboxylate 73 (see Scheme 7).
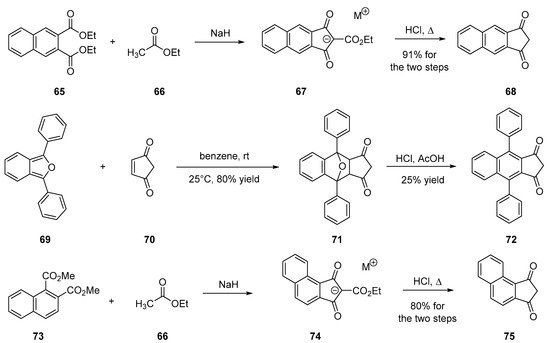
Scheme 7. Synthetic routes to 68, 72 and 75.
2.3.2. Halogenated Indane-1,3-Diones
Halogenation of indane-1,3-dione derivatives subsequent to their synthesis is not possible such that such derivatives can only be obtained by first introducing halogens onto their corresponding precursors. Notably, as a first synthetic approach, halogenated phthalic anhydrides were converted as indane-1,3-diones using ethyl acetoacetate, and a series of halogenated indane-1,3-diones 76–82 is presented in Scheme 8 [20][21][28][37][38][40][61]. Parallel to this, the strategy previously mentioned that AlCl3-promoted acylation of a benzoyl chloride derivative (83) with malonyl chloride 24 proved to be another effective approach to design chlorinated indane-1,3-dione derivatives (84) (see Scheme 8).
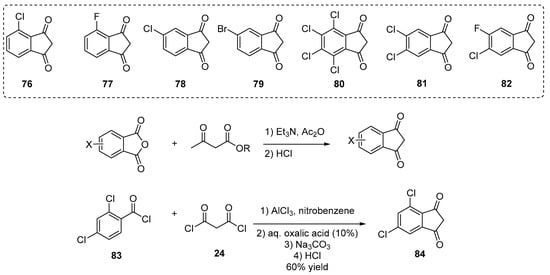
Scheme 8. Examples of halogenated indane-1,3-diones 76–82, 84 reported in the literature.
2.3.3. Introduction of Various Electron-Withdrawing Groups on Aromatic Ring
Nitration
As previously mentioned for halogenation, electrophilic aromatic substitution cannot be carried out on indane-1,3-dione 4 such that a post-functionalization with nitro groups is required. To date, only few indane-1,3-diones bearing nitro groups have been reported in the literature (see Scheme 9) [21][22][62].
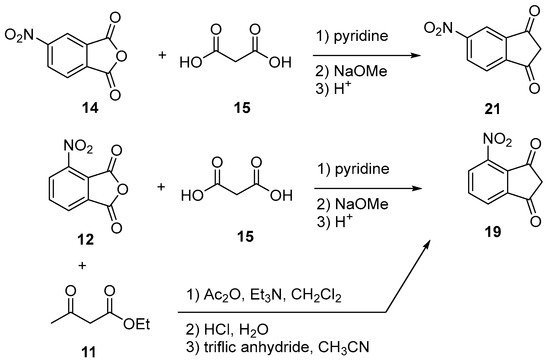
Scheme 9. Synthesis of nitro-substituted indane-1,3-diones 19 and 21.
2.3.4. Cyanation
To the best of researchers' knowledge, no cyano-substituted indane-1,3-dione derivatives have been reported to date. Furthermore, such acceptors are as crucial as the cyano groups, which are among the best electron-accepting groups.
2.3.5. Introduction of Alkoxy-Carbonyl Groups
Here again, only the post-functionalization of naphthalic anhydrides was used to introduce CO2R groups. An example is provided below with 32 (see Scheme 10) [31]. By using ethyl acetoacetate 11, anhydride acetic as the solvent and triethylamine as the base, 32 could be obtained from 32a in 71% yield.
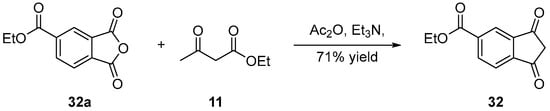
Scheme 10. Ethoxy-carbonyl compound 32.
2.4. Chemical Engineering around the Methylene Group
2.4.1. Knoevenagel Reaction
Due to the presence of the two ketones groups on both sides of the methylene groups, indane-1,3-dione 4 possesses a privileged group for realizing Knoevenagel reactions. In the case of indane-1,3-dione 4 and its substituted derivatives, the condensation reaction can be carried out in the conditions initially used by Knoevenagel in 1894 to condense benzaldehyde with ethyl acetoacetate 11, namely in ethanol with a catalytic amount of piperidine [61]. Typically, Knoevenagel reactions performed with 4 or 68 can be realized with reaction yields higher than 70% (see Table 1 and Scheme 11) [12]. As the main interest of this reaction, use of a highly polar solvent favors the precipitation of dyes 95–114 upon cooling, and the reaction can be carried out in green conditions since a non-dangerous solvent can be used. Additionally, the work-up can be limited to a simple filtration, avoiding the use of complicated purification processes [56][57].
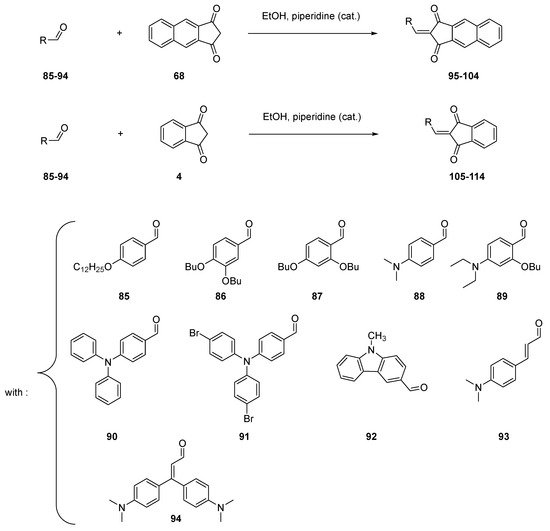
Scheme 11. Synthesis of push–pull dyes 95–114.
Table 1. Reaction yields obtained for the synthesis of 95–114.
| compounds | 95 | 96 | 97 | 98 | 99 | 100 | 101 | 102 | 103 | 104 |
| reaction yields | 88 | 84 | 88 | 74 | 94 | 89 | 92 | 85 | 84 | 88 |
| compounds | 105 | 106 | 107 | 108 | 109 | 110 | 111 | 112 | 113 | 114 |
| reaction yields | 74 | 85 | 75 | 82 | 92 | 87 | 81 | 78 | 89 | 85 |
When 2-(3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile 28 and its analogues are involved in Knoevenagel reactions, another amine should be used, and diisopropylethylamine (DIPEA), which is a none-nucleophilic base, is the most popular one. As reported in several recent works, an unexpected nucleophilic addition of secondary amines onto the cyano groups of the push–pull dyes can occur, giving rise to a cyclization reaction and producing 3-(dialkylamino)-1,2-dihydro-9-oxo-9H-indeno [2,1-c]pyridine-4-carbonitrile derivatives 115, according to the mechanism proposed in Scheme 12.
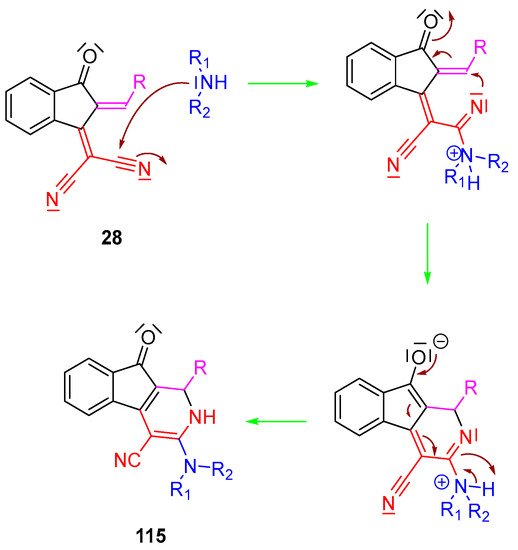
Scheme 12. Mechanism resulting in the synthesis of 3-(dialkylamino)-1,2-dihydro-9-oxo-9H-indeno [2,1-c]pyridine-4-carbonitrile derivatives 115.
Numerous examples of undesired cyclization reactions have notably been reported with piperidine providing 3-(dialkylamino)-1,2-dihydro-9-oxo-9H-indeno [2,1-c]pyridine-4-carbonitrile derivatives 115 instead of the expected push–pull dyes [62][63][64]. In 2019, an unprecedented nucleophilic addition of piperidine on 101 was also reported, providing 102 after dehydration. The mechanism supporting the formation of this unexpected structure is depicted in Scheme 13.
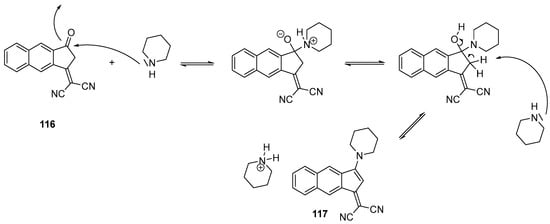
Scheme 13. Mechanism supporting the formation of 117 starting from 116.
When 2,2′-(1H-indene-1,3(2H)-diylidene)dimalononitrile 29 and its derivatives are engaged in Knoevenagel reactions, the high stability of their anions in basic conditions impedes the Knoevenagel reactions to proceed. Therefore, acidic conditions should be used, and acetic anhydride is commonly used in this aim (see Scheme 14) [65][66].
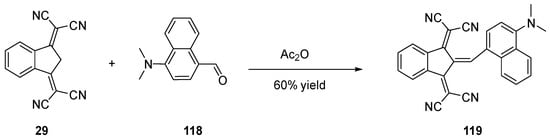
Scheme 14. Synthetic routes to 119 starting from 29.
Besides, several reports mention the use of the classical piperidine/ethanol conditions to condense 29 onto aromatic aldehydes, despites the strong deactivation of the 29 anion in basic conditions. A few examples of products obtained in these conditions are presented in Scheme 15 (121 [67], 123 [67] or 125 [68]).
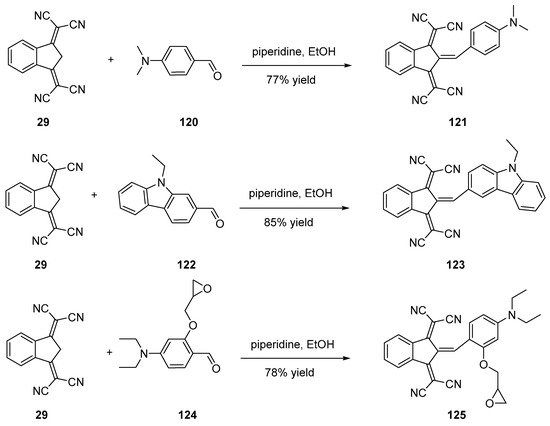
Scheme 15. Knoevenagel reactions performed in classical ethanol/piperidine conditions.
To avoid the use of base, several authors replaced ethanol by solvents of higher boiling points such as methylethylketone (see Scheme 16) [69]. By refluxing 29 and 126 at elevated temperature, 127 could be obtained in 42% yield.
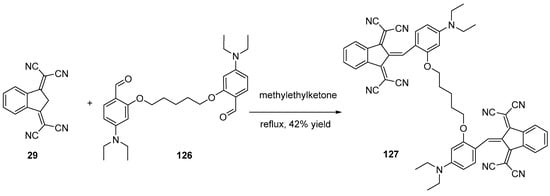
Scheme 16. Synthetic route to push–pull dye 127.
2.4.2. Oxidation Reaction
Among indane-1,3-dione derivatives, ninhydrin is well known, as it can be advantageously used as a revelator for thin layer chromatography (TLC). Over the years, several strategies have been developed to access to these structures [70]. For instance, oxidation of indane-1-one 128 with selenium oxide (SeO2) can furnish ninhydrin 138 [71], but this reaction was not limited to 4, and the oxidation reaction tolerates various substituents such as OMe, tert-Bu, Me, Br, CF3 or NO2 (see compounds 139–147, Scheme 17) [72]. Direct oxidation of 4 was also investigated to convert it, as 138 and ninhydrin 138 could be prepared in 48% yield using the dual oxidizing system SeO2/H2O2 [73] in 40% yield for the two steps using iodobenzene diacetate [74] and 94% yield using N-bromosuccinimide (NBS) in DMSO [75]. However, all attempts to oxidize 4 as 138 using sodium hypochlorite failed, and phthalic acid was isolated as the unique product of the reaction [76]. The authors also demonstrated 138 to be oxidized as phthalic acid, evidencing that sodium hypochlorite is a too strong oxidant. The oxidation reaction with N-bromosuccinimide (NBS) in DMSO was not limited to indane-1,3-dione 4, and indane-1-one 149 and indane-2-one 128 could also be oxidized using the same procedure, providing 138 in 82 and 85% yield, respectively. Photooxidation of 4 in the presence of tetrabutylammonium hexafluorophosphate and oxygen using Rose Bengal as the photosensitizer could convert 4 as 138 in 75% yield upon irradiation with a UV light [77].
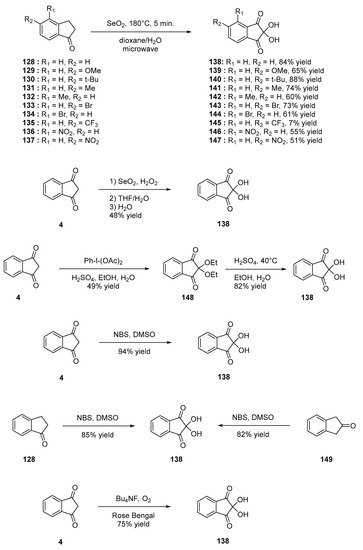
Scheme 17. Synthetic route to ninhydrin 138.
2.4.3. Halogenation
α,α-Dihalogenation reactions of carbonyl compounds have been extensively studied in the literature [78][79][80][81][82][83][84][85][86][87][88], and different procedures were thus developed for the halogenation of indane-1,3-dione 4. Notably, traditional reagents of halogenation including N-chlorosuccinimide (NCS) or NBS in ethanol could provide 150 and 151 in 95% and 92% yields [89]. Several green syntheses were also developed, all based on mechanical ball milling. Using this approach, halogenating agents such as trichloroisocyanuric acid or tribromoisocyanuric acid could furnish 150 and 151 in 98 and 97% yields [90]. Similarly, mechanosynthesis of 150 could be efficiently achieved by employing sodium bromide and oxone (98% yield) [85] or ammonium bromide and oxone (see Scheme 18) [91].
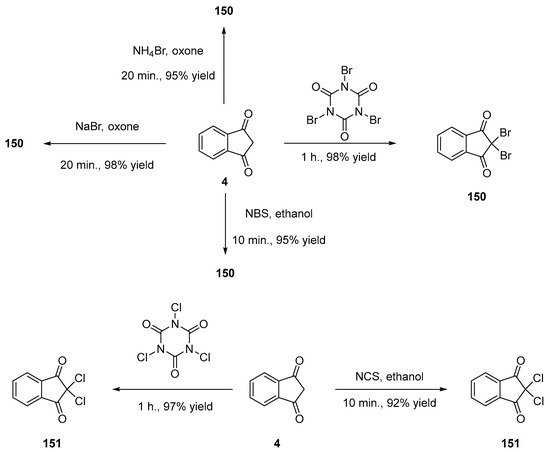
Scheme 18. Different routes for halogenation of indane-1,3-dione 4 at the methylene position.
The conversion of nucleophilic halogens to electrophilic ones could be realized by reacting Lewis acids such as ZnBr2 or AlCl3 with lead tetraacetate [92]. High reaction yields were also obtained during the synthesis of 150 while using KBr/KBrO3 (86% yield) [93], 1,3-dibromo-5,5-dimethylhydantoin in acetic acid (88% yield) [94]. Similarly, 151 could be obtained while reacting 4 with 1,3-dichloro-5,5-dimethylhydantoin in acetic acid (89% yield) [94]. Finally, selectfluor® (1-chloromethyl-4-fluoro-1,4-diazoniabicyclo [2.2.2]octane bis(tetrafluoroborate)) was the most widely studied fluorinated agent for fluorination of 4 in water while using a surfactant (Genapol LRO) (74% yield) [95], sodium dodecyl sulfate in water (93% yield) [96], or acetonitrile as solvent (60% yield) (see Scheme 19) [41][97]. Furthermore, numerous drawbacks concerning the electrophilic fluorination with selectfluor® were reported in the literature. Notably, parallel to the formation of the expected F+ cation, formation of radical species (F•) by single electron transfer has also been proposed, even if the mechanistic studies have not fully elucidated the mechanism. Nevertheless, formation of an intermediate monofluorination state could be demonstrated [98].
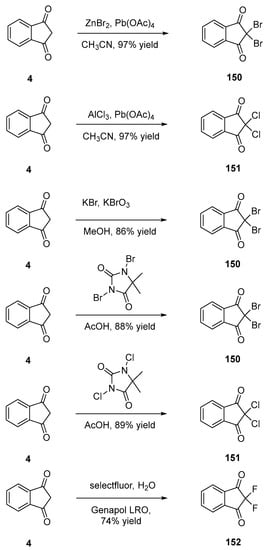
Scheme 19. Fluorination reactions of indane-1,3-dione 4.
In the case of indane-1,3-dione 4, fluorination was proposed as occurring by means of an attack of the double bond of enol onto selectfluor®, followed by a deprotonation with the resulting diazoniabicyclo [2.2.2]octane species. By iterating the reaction a second time, 152 could be obtained (see Scheme 20) [97]. Considering that there is still a lack of efficiency for the α,α-dibromination of 1,3-diketones, the photoredox catalysis was envisioned as a possible alternative to conventional chemistry to improve the selectivity during bromination [99][100][101][102]. Light is also a traceless reagent so that light-promoted chemistry perfectly fits with the concepts of green chemistry. For bromination, light-activated radical reactions are also extensively described in the literature, enabling to efficiently generate bromine radicals.
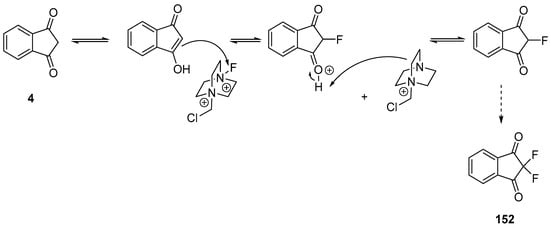
Scheme 20. Mechanism involved in the fluorination of 4.
In 2019, an interesting reaction was developed to accelerate the formation of bromide radicals and, in this aim, N-bromosuccinimide (NBS) was irradiated in the presence of bromoacetic acid and under visible light [103]. By this unique combination, two concurrent bromination mechanisms could be evidenced, the first one consisting in an electrophilic bromination resulting from the photoassisted formation of bromine, and the second one consisting in a radical bromination process resulting from the formation of bromide radicals promoted by light (see Scheme 21).
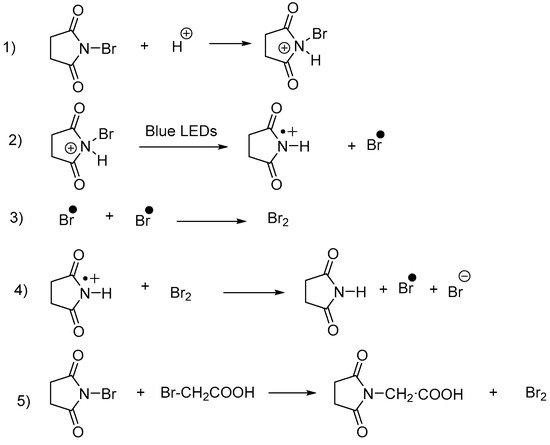
Scheme 21. Mechanism supporting the coexistence of a radical and an electrophilic bromination pathway for the bromination of 4.
By the presence of these double sources of brominating agents, the exceptional reaction yield of 99% could be obtained as well as accounts from the coexistence of both the electrophilic and the radical bromination reactions (see Scheme 22).
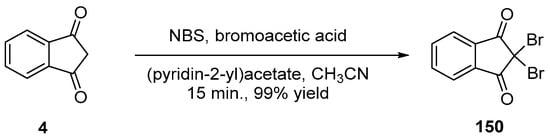
Scheme 22. Synthesis of 150 using NBS as the bromination agent.
2.4.4. Cyanation
In 1977, an interesting procedure was developed to introduce a cyano group at the methylene position of indane-1,3-dione. Inspired by the synthesis of indane-1,3-dione 4 starting from dialkyl phthalate and ethyl acetate, an analogue procedure was proposed, where ethyl acetate 51 was replaced by acetonitrile (see Scheme 23) [104]. Using sodium methanoate as the base, 153 could be obtained in 78% yield.
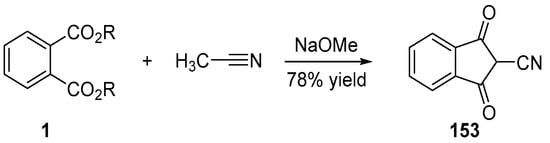
Scheme 23. Synthetic route to 153.
2.4.5. Nitration
Nitration of the methylene group of indane-1,3-dione 4 can be performed in one step, by using nitric acid as the reagent [105]. Furthermore, 154 could be obtained in 78% yield (see Scheme 24).
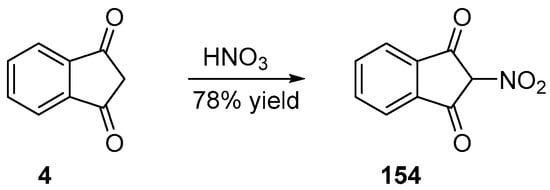
Scheme 24. Synthetic route to 154.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27185976
References
- Patil, S.A.; Patil, R.; Patil, S.A. Recent Developments in Biological Activities of Indanones. Eur. J. Med. Chem. 2017, 138, 182–198.
- Turek, M.; Szczęsna, D.; Koprowski, M.; Bałczewski, P. Synthesis of 1-Indanones with a Broad Range of Biological Activity. Beilstein J. Org. Chem. 2017, 13, 451–494.
- Menezes, J.C.J.M.D.S. Arylidene Indanone Scaffold: Medicinal Chemistry and Structure–Activity Relationship View. RSC Adv. 2017, 7, 9357–9372.
- Costanzo, P.; Cariati, L.; Desiderio, D.; Sgammato, R.; Lamberti, A.; Arcone, R.; Salerno, R.; Nardi, M.; Masullo, M.; Oliverio, M. Design, Synthesis, and Evaluation of Donepezil-Like Compounds as AChE and BACE-1 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 470–475.
- Vacca, J.P.; Dorsey, B.D.; Schleif, W.A.; Levin, R.B.; McDaniel, S.L.; Darke, P.L.; Zugay, J.; Quintero, J.C.; Blahy, O.M.; Roth, E. L-735,524: An Orally Bioavailable Human Immunodeficiency Virus Type 1 Protease Inhibitor. Proc. Natl. Acad. Sci. USA 1994, 91, 4096.
- Luo, H.-F.; Zhang, L.-P.; Hu, C.-Q. Five Novel Oligostilbenes from the Roots of Caragana Sinica. Tetrahedron 2001, 57, 4849–4854.
- Nagle, D.G.; Zhou, Y.-D.; Park, P.U.; Paul, V.J.; Rajbhandari, I.; Duncan, C.J.G.; Pasco, D.S. A New Indanone from the Marine Cyanobacterium Lyngbya Majuscula That Inhibits Hypoxia-Induced Activation of the VEGF Promoter in Hep3B Cells. J. Nat. Prod. 2000, 63, 1431–1433.
- Kim, J.; Kim, I. Design and Synthesis of a Hybrid Framework of Indanone and Chromane: Total Synthesis of a Homoisoflavanoid, Brazilane. Org. Biomol. Chem. 2018, 16, 89–100.
- Yang, Y.; Philips, D.; Pan, S. A Concise Synthesis of Paucifloral F and Related Indanone Analogues via Palladium-Catalyzed α-Arylation. J. Org. Chem. 2011, 76, 1902–1905.
- Buckingham, J. Dictionary of Natural Products, Supplement 1, 1st ed.; Taylor & Francis: New York, NY, USA, 1994; ISBN 978-1-00-305992-9.
- Winzenberg, K.N.; Kemppinen, P.; Scholes, F.H.; Collis, G.E.; Shu, Y.; Birendra Singh, T.; Bilic, A.; Forsyth, C.M.; Watkins, S.E. Indan-1,3-Dione Electron-Acceptor Small Molecules for Solution-Processable Solar Cells: A Structure–Property Correlation. Chem. Commun. 2013, 49, 6307–6309.
- Pigot, C.; Noirbent, G.; Bui, T.-T.; Péralta, S.; Gigmes, D.; Nechab, M.; Dumur, F. Push-Pull Chromophores Based on the Naphthalene Scaffold: Potential Candidates for Optoelectronic Applications. Materials 2019, 12, 1342.
- Patel, A.; Giles, D.; Basavarajaswamy, G.; Sreedhar, C.; Patel, A. Synthesis, Pharmacological Evaluation and Molecular Docking Studies of Indanone Derivatives. Med. Chem. Res. 2012, 21, 4403–4411.
- Karthik, R.; Jasmin Sajni, R.; Sasikumar, S.; Kalyan, S.B.; Christina, A.J.M.; Jagan, A.; Sundara Saravanan, K. Evaluation of Anti-Tubercular Activity of Some Synthesised Benz Spiro-Oxirane Derivatives of Indane-1,3-Dione. Pharmacologyonline 2008, 2, 176–191.
- Liu, J.; Hu, K.-F.; Qu, J.-P.; Kang, Y.-B. Organopromoted Selectivity-Switchable Synthesis of Polyketones. Org. Lett. 2017, 19, 5593–5596.
- Mardani, H.R.; Golchoubian, H. Selective and Efficient C-H Oxidation of Alkanes with Hydrogen Peroxide Catalyzed by a Manganese(III) Schiff Base Complex. J. Mol. Catal. Chem. 2006, 259, 197–200.
- Delaval, N.; Bouquillon, S.; Hénin, F.; Muzart, J. Use of Benzotrifluoride as Solvent for Chromium-Catalysed Oxidations with Sodium Percarbonate. J. Chem. Res. Synop. 1999, 4, 286–287.
- He, G.; Wu, C.; Zhou, J.; Yang, Q.; Zhang, C.; Zhou, Y.; Zhang, H.; Liu, H. A Method for Synthesis of 3-Hydroxy-1-Indanones via Cu-Catalyzed Intramolecular Annulation Reactions. J. Org. Chem. 2018, 83, 13356–13362.
- Kumar, K.; Kumar, P.; Joshi, P.; Rawat, D.S. IBX-TfOH Mediated Oxidation of Alcohols to Aldehydes and Ketones under Mild Reaction Conditions. Tetrahedron Lett. 2020, 61, 151749.
- Marvi, O.; Giahi, M. Montmorillonite KSF Clay as Novel and Recyclable Heterogeneous Catalyst for the Microwave Mediated Synthesis of Indan-1,3-Diones. Bull. Korean Chem. Soc. 2009, 30, 2918–2920.
- Guo, S.; Zhang, N.; Tang, X.; Mao, Z.; Zhang, X.; Yan, M.; Xuan, Y. Cyclopropanation of Active Methylene Compounds with β-Alkoxycarbonyl Vinylsulfonium Salts. Chin. Chem. Lett. 2019, 30, 406–408.
- Berezina, G.R.; Vorob’ev, Y.G.; Mukhanova, O.N. Macroheterocyclic Compounds Based on Nitro- and Chloro-1,3-Indandiones. Russ. J. Gen. Chem. 2005, 75, 1594–1598.
- Li, S.; Ye, L.; Zhao, W.; Zhang, S.; Mukherjee, S.; Ade, H.; Hou, J. Energy-Level Modulation of Small-Molecule Electron Acceptors to Achieve over 12% Efficiency in Polymer Solar Cells. Adv. Mater. 2016, 28, 9423–9429.
- Cui, Y.; Ren, H.; Yu, J.; Wang, Z.; Qian, G. An Indanone-Based Alkoxysilane Dye with Second Order Nonlinear Optical Properties. Dyes Pigments 2009, 81, 53–57.
- Abdelrazek, F.M.; Metz, P.; Jaeger, A. Some Reactions with Indane-1,3-Dione: A Facile Synthesis of Pentacycline Heterocyclic Analogues. J. Heterocycl. Chem. 2019, 56, 1939–1945.
- Tirelli, N.; Amabile, S.; Cellai, C.; Pucci, A.; Regoli, L.; Ruggeri, G.; Ciardelli, F. New Terthiophene Derivatives for Ultrahigh Molecular Weight Polyethylene-Based Absorption Polarizers. Macromolecules 2001, 34, 2129–2137.
- Shang, Y.; Wen, Y.; Li, S.; Du, S.; He, X.; Cai, L.; Li, Y.; Yang, L.; Gao, H.; Song, Y. A Triphenylamine-Containing Donor−Acceptor Molecule for Stable, Reversible, Ultrahigh Density Data Storage. J. Am. Chem. Soc. 2007, 129, 11674–11675.
- Planells, M.; Robertson, N. Naphthyl Derivatives Functionalised with Electron Acceptor Units–Synthesis, Electronic Characterisation and DFT Calculations. Eur. J. Org. Chem. 2012, 2012, 4947–4953.
- Yang, X.; Fox, T.; Berke, H. Synthetic and Mechanistic Studies of Metal-Free Transfer Hydrogenations Applying Polarized Olefins as Hydrogen Acceptors and Amine Borane Adducts as Hydrogen Donors. Org. Biomol. Chem. 2012, 10, 852–860.
- Morales, A.R.; Frazer, A.; Woodward, A.W.; Ahn-White, H.-Y.; Fonari, A.; Tongwa, P.; Timofeeva, T.; Belfield, K.D. Design, Synthesis, and Structural and Spectroscopic Studies of Push–Pull Two-Photon Absorbing Chromophores with Acceptor Groups of Varying Strength. J. Org. Chem. 2013, 78, 1014–1025.
- Matsui, M.; Tanaka, N.; Funabiki, K.; Haishima, Y.; Manseki, K.; Jin, J.; Inoue, Y.; Higashijima, S.; Kubota, Y. Application of Indoline Dyes Attached with Strongly Electron-Withdrawing Carboxylated Indan-1,3-Dione Analogues Linked with a Hexylthiophene Ring to Dye-Sensitized Solar Cells. Tetrahedron 2018, 74, 3498–3506.
- Dai, S.; Zhao, F.; Zhang, Q.; Lau, T.-K.; Li, T.; Liu, K.; Ling, Q.; Wang, C.; Lu, X.; You, W.; et al. Fused Nonacyclic Electron Acceptors for Efficient Polymer Solar Cells. J. Am. Chem. Soc. 2017, 139, 1336–1343.
- Zhu, J.; Li, S.; Liu, X.; Yao, H.; Wang, F.; Zhang, S.; Sun, M.; Hou, J. Subtle Side-Chain Tuning on Terminal Groups of Small Molecule Electron Acceptors for Efficient Fullerene-Free Polymer Solar Cells. J. Mater. Chem. A 2017, 5, 15175–15182.
- Lai, H.; Chen, H.; Zhou, J.; Qu, J.; Wang, M.; Xie, W.; Xie, Z.; He, F. 3D Interpenetrating Network for High-Performance Nonfullerene Acceptors via Asymmetric Chlorine Substitution. J. Phys. Chem. Lett. 2019, 10, 4737–4743.
- Aldrich, T.J.; Matta, M.; Zhu, W.; Swick, S.M.; Stern, C.L.; Schatz, G.C.; Facchetti, A.; Melkonyan, F.S.; Marks, T.J. Fluorination Effects on Indacenodithienothiophene Acceptor Packing and Electronic Structure, End-Group Redistribution, and Solar Cell Photovoltaic Response. J. Am. Chem. Soc. 2019, 141, 3274–3287.
- Swick, S.M.; Zhu, W.; Matta, M.; Aldrich, T.J.; Harbuzaru, A.; Lopez Navarrete, J.T.; Ponce Ortiz, R.; Kohlstedt, K.L.; Schatz, G.C.; Facchetti, A.; et al. Closely Packed, Low Reorganization Energy π-Extended Postfullerene Acceptors for Efficient Polymer Solar Cells. Proc. Natl. Acad. Sci. USA 2018, 115, E8341.
- Cui, Y.; Yang, C.; Yao, H.; Zhu, J.; Wang, Y.; Jia, G.; Gao, F.; Hou, J. Efficient Semitransparent Organic Solar Cells with Tunable Color Enabled by an Ultralow-Bandgap Nonfullerene Acceptor. Adv. Mater. 2017, 29, 1703080.
- Yao, H.; Cui, Y.; Yu, R.; Gao, B.; Zhang, H.; Hou, J. Design, Synthesis, and Photovoltaic Characterization of a Small Molecular Acceptor with an Ultra-Narrow Band Gap. Angew. Chem. Int. Ed. 2017, 56, 3045–3049.
- Bürckstümmer, H.; Tulyakova, E.V.; Deppisch, M.; Lenze, M.R.; Kronenberg, N.M.; Gsänger, M.; Stolte, M.; Meerholz, K.; Würthner, F. Efficient Solution-Processed Bulk Heterojunction Solar Cells by Antiparallel Supramolecular Arrangement of Dipolar Donor–Acceptor Dyes. Angew. Chem. Int. Ed. 2011, 50, 11628–11632.
- Wilbuer, J.; Schnakenburg, G.; Esser, B. Syntheses, Structures and Optoelectronic Properties of Spiroconjugated Cyclic Ketones. Eur. J. Org. Chem. 2016, 2016, 2404–2412.
- Sloop, J.C.; Boyle, P.D.; Fountain, A.W.; Gomez, C.; Jackson, J.L.; Pearman, W.F.; Schmidt, R.D.; Weyand, J. Novel Fluorinated Indanone, Tetralone and Naphthone Derivatives: Synthesis and Unique Structural Features. Appl. Sci. 2012, 2, 61–99.
- Nikulin, V.I.; Lugovskaya, N.Y.; Sveshnikov, N.N.; Pisarenko, L.M.; D’yachkovskaya, R.F. Synthesis and Antitumor Activity of 2-Arylindane-1,3-Dione Derivatives with an Alkylating Fragment. Pharm. Chem. J. 1994, 28, 146–149.
- Abdel-Latif, F.F.; Mashaly, M.M.; El-Gawish, E.H. ChemInform Abstract: Synthesis of Heterocycles Through Reactions of Nucleophiles with Acrylonitriles. Part 15. Synthesis of Some New Functionalized Benzo(b) Pyrans and Indeno(1,2-b)Pyrans of Potential Biological Activity. ChemInform 1995, 26, 42.
- Becker, H.-D. 2--1, 3-Indanedione and Its Tautomer. U.S. Patent 3,356,732, 25 December 1967.
- Liu, Y.; Saldivar, A.; Bess, J.; Solomon, L.; Chen, C.-M.; Tripathi, R.; Barrett, L.; Richardson, P.L.; Molla, A.; Kohlbrenner, W.; et al. Investigating the Origin of the Slow-Binding Inhibition of HCV NS3 Serine Protease by a Novel Substrate Based Inhibitor. Biochemistry 2003, 42, 8862–8869.
- Siddiqui, N.; Ahuja, P.; Ahsan, W.; Pandeya, S.N.; Shamsher Alam, M. Thiadiazoles: Progress Report on Biological Activities. J. Chem. Pharm. Res. 2009, 1, 19–30.
- Salgın-Gökşen, U.; Gökhan-Kelekçi, N.; Göktaş, Ö.; Köysal, Y.; Kılıç, E.; Işık, Ş.; Aktay, G.; Özalp, M. 1-Acylthiosemicarbazides, 1,2,4-Triazole-5(4H)-Thiones, 1,3,4-Thiadiazoles and Hydrazones Containing 5-Methyl-2-Benzoxazolinones: Synthesis, Analgesic-Anti-Inflammatory and Antimicrobial Activities. Bioorg. Med. Chem. 2007, 15, 5738–5751.
- Schenone, S.; Brullo, C.; Bruno, O.; Bondavalli, F.; Ranise, A.; Filippelli, W.; Rinaldi, B.; Capuano, A.; Falcone, G. New 1,3,4-Thiadiazole Derivatives Endowed with Analgesic and Anti-Inflammatory Activities. Bioorg. Med. Chem. 2006, 14, 1698–1705.
- Wei, M.-X.; Feng, L.; Li, X.-Q.; Zhou, X.-Z.; Shao, Z.-H. Synthesis of New Chiral 2,5-Disubstituted 1,3,4-Thiadiazoles Possessing γ-Butenolide Moiety and Preliminary Evaluation of in Vitro Anticancer Activity. Eur. J. Med. Chem. 2009, 44, 3340–3344.
- Mavrova, A.T.; Wesselinova, D.; Tsenov, Y.A.; Denkova, P. Synthesis, Cytotoxicity and Effects of Some 1,2,4-Triazole and 1,3,4-Thiadiazole Derivatives on Immunocompetent Cells. Eur. J. Med. Chem. 2009, 44, 63–69.
- Siddiqui, N.; Ali, S.; Khan, S.; Drabu, S.; Rana, A.; Alam, M. Synthesis of 3-Arylamino-4-Aryl-5-(N-Arylthiocarbonylimino)-4, 5-Dihydro-1, 2, 4-Thiadiazoles as Anticonvulsant Agents. Indian J. Heterocycl. Chem. 2004, 14, 159–160.
- Kuş, C.; Ayhan-Kılcıgil, G.; Özbey, S.; Kaynak, F.B.; Kaya, M.; Çoban, T.; Can-Eke, B. Synthesis and Antioxidant Properties of Novel N-Methyl-1,3,4-Thiadiazol-2-Amine and 4-Methyl-2H-1,2,4-Triazole-3(4H)-Thione Derivatives of Benzimidazole Class. Bioorg. Med. Chem. 2008, 16, 4294–4303.
- Cressier, D.; Prouillac, C.; Hernandez, P.; Amourette, C.; Diserbo, M.; Lion, C.; Rima, G. Synthesis, Antioxidant Properties and Radioprotective Effects of New Benzothiazoles and Thiadiazoles. Bioorg. Med. Chem. 2009, 17, 5275–5284.
- Sharma, R.; Misra, G.P.; Sainy, J.; Chaturvedi, S.C. Synthesis and Biological Evaluation of 2-Amino-5-Sulfanyl-1,3,4-Thiadiazole Derivatives as Antidepressant, Anxiolytics and Anticonvulsant Agents. Med. Chem. Res. 2011, 20, 245–253.
- Muhammad, Z.A.; Masaret, G.S.; Amin, M.M.; Abdallah, M.A.; Farghaly, T.A. Anti-Inflammatory, Analgesic and Anti-Ulcerogenic Activities of Novel Bis-Thiadiazoles, Bis-Thiazoles and Bis-Formazanes. Med. Chem. 2017, 13, 226–238.
- Pigot, C.; Noirbent, G.; Peralta, S.; Duval, S.; Nechab, M.; Gigmes, D.; Dumur, F. Unprecedented Nucleophilic Attack of Piperidine on the Electron Acceptor during the Synthesis of Push-Pull Dyes by a Knoevenagel Reaction. Helv. Chim. Acta 2019, 102, e1900229.
- Pigot, C.; Noirbent, G.; Peralta, S.; Duval, S.; Bui, T.-T.; Aubert, P.-H.; Nechab, M.; Gigmes, D.; Dumur, F. New Push-Pull Dyes Based on 2-(3-Oxo-2,3-Dihydro-1H-CyclopentaNaphthalen-1-Ylidene)Malononitrile: An Amine-Directed Synthesis. Dyes Pigments 2020, 175, 108182.
- Feng, H.; Qiu, N.; Wang, X.; Wang, Y.; Kan, B.; Wan, X.; Zhang, M.; Xia, A.; Li, C.; Liu, F.; et al. An A-D-A Type Small-Molecule Electron Acceptor with End-Extended Conjugation for High Performance Organic Solar Cells. Chem. Mater. 2017, 29, 7908–7917.
- Li, R.; Liu, G.; Xiao, M.; Yang, X.; Liu, X.; Wang, Z.; Ying, L.; Huang, F.; Cao, Y. Non-Fullerene Acceptors Based on Fused-Ring Oligomers for Efficient Polymer Solar Cells via Complementary Light-Absorption. J. Mater. Chem. A 2017, 5, 23926–23936.
- Sanguinet, L.; Williams, J.C.; Yang, Z.; Twieg, R.J.; Mao, G.; Singer, K.D.; Wiggers, G.; Petschek, R.G. Synthesis and Characterization of New Truxenones for Nonlinear Optical Applications. Chem. Mater. 2006, 18, 4259–4269.
- Knoevenagel, E. Ueber Eine Darstellungsweise Des Benzylidenacetessigesters. Berichte Dtsch. Chem. Ges. 1896, 29, 172–174.
- Landmesser, T.; Linden, A.; Hansen, H.-J. A Novel Route to 1-Substituted 3-(Dialkylamino)-9-Oxo-9H-IndenoPyridine-4-Carbonitriles. Helv. Chim. Acta 2008, 91, 265–284.
- Helmy, S.; Oh, S.; Leibfarth, F.A.; Hawker, C.J.; Read de Alaniz, J. Design and Synthesis of Donor–Acceptor Stenhouse Adducts: A Visible Light Photoswitch Derived from Furfural. J. Org. Chem. 2014, 79, 11316–11329.
- Gao, M.; Su, H.; Lin, Y.; Ling, X.; Li, S.; Qin, A.; Tang, B.Z. Photoactivatable Aggregation-Induced Emission Probes for Lipid Droplets-Specific Live Cell Imaging. Chem. Sci. 2017, 8, 1763–1768.
- Capobianco, A.; Esposito, A.; Caruso, T.; Borbone, F.; Carella, A.; Centore, R.; Peluso, A. Tuning Wavefunction Mixing in Push–Pull Molecules: From Neutral to Zwitterionic Compounds. Eur. J. Org. Chem. 2012, 2012, 2980–2989.
- Bello, K.A.; Cheng, L.; Griffiths, J. Near-Infrared Absorbing Methine Dyes Based on Dicyanovinyl Derivatives of Indane-1,3-Dione. J. Chem. Soc. Perkin Trans. 1987, 2, 815–818.
- Singh, A.; Lim, C.-K.; Lee, Y.-D.; Maeng, J.; Lee, S.; Koh, J.; Kim, S. Tuning Solid-State Fluorescence to the Near-Infrared: A Combinatorial Approach to Discovering Molecular Nanoprobes for Biomedical Imaging. ACS Appl. Mater. Interfaces 2013, 5, 8881–8888.
- Zilinskaite, V.; Gudeika, D.; Grazulevicius, J.V.; Hladka, I. Synthesis and Cationic Polymerization of Oxyranyl-Functionalized Indandiones. Polym. Bull. 2016, 1, 229–239.
- Gudeika, D.; Zilinskaite, V.; Grazulevicius, J.V.; Lytvyn, R.; Rutkis, M.; Tokmakov, A. 4-(Diethylamino)Salicylaldehyde-Based Twin Compounds as NLO-Active Materials. Dyes Pigments 2016, 134, 244–250.
- Ziarani, G.M.; Lashgari, N.; Azimian, F.; Kruger, H.G.; Gholamzadeh, P. Ninhydrin in Synthesis of Heterocyclic Compounds. ARKIVOC 2015, 2015, 1–139.
- Marminon, C.; Nacereddine, A.; Bouaziz, Z.; Nebois, P.; Jose, J.; Le Borgne, M. Microwave-Assisted Oxidation of Indan-1-Ones into Ninhydrins. Tetrahedron Lett. 2015, 56, 1840–1842.
- Jong, J.A.W.; Moret, M.-E.; Verhaar, M.C.; Hennink, W.E.; Gerritsen, K.G.F.; van Nostrum, C.F. Effect of Substituents on the Reactivity of Ninhydrin with Urea. ChemistrySelect 2018, 3, 1224–1229.
- Taherpour, A.; Kamal, S. 1,2,3-Trione Compounds Synthesis by Oxidation 1,3-Diketones. Asian J. Chem. 2007, 19, 4107–4109.
- Prakash, O.; Sharma, P.K.; Saini, N. ChemInform Abstract: A New Synthesis of Ninhydrin and Its Ketals Using Iodobenzene Diacetate. ChemInform 1995, 26, 39.
- Tatsugi, J.; Izawa, Y. A Facile One-Pot Synthesis of Vicinal Di- and Tri-Ketones from α-Methylene Ketones by N-Bromosuccinimide-Dimethyl Sulphoxide Oxidation. J. Chem. Res. Synop. Print 1988, 11, 356–357.
- Vickery, B.; Kaberia, F. Reactions of Sodium Hypochlorite with Some Compounds Having Reactive Methylene Groups. Experientia 1979, 35, 299.
- Wasserman, H.H.; Pickett, J.E. The Fluoride Ion Effect in the Reactions of Singlet Oxygen with Enols. Tetrahedron 1985, 41, 2155–2162.
- Gao, S.; Bethel, T.K.; Kakeshpour, T.; Hubbell, G.E.; Jackson, J.E.; Tepe, J.J. Substrate Controlled Regioselective Bromination of Acylated Pyrroles Using Tetrabutylammonium Tribromide (TBABr3). J. Org. Chem. 2018, 83, 9250–9255.
- Wengryniuk, S.E.; Weickgenannt, A.; Reiher, C.; Strotman, N.A.; Chen, K.; Eastgate, M.D.; Baran, P.S. Regioselective Bromination of Fused Heterocyclic N-Oxides. Org. Lett. 2013, 15, 792–795.
- Pathak, T.P.; Miller, S.J. Site-Selective Bromination of Vancomycin. J. Am. Chem. Soc. 2012, 134, 6120–6123.
- Shi, X.; Dai, L. Mild Halogenation of Stabilized Ester Enolates by Cupric Halides. J. Org. Chem. 1993, 58, 4596–4598.
- Yang, D.; Yan, Y.-L.; Lui, B. Mild α-Halogenation Reactions of 1,3-Dicarbonyl Compounds Catalyzed by Lewis Acids. J. Org. Chem. 2002, 67, 7429–7431.
- Tanemura, K.; Suzuki, T.; Nishida, Y.; Satsumabayashi, K.; Horaguchi, T. A Mild and Efficient Procedure for α-Bromination of Ketones Using N-Bromosuccinimide Catalysed by Ammonium Acetate. Chem. Commun. 2004, 4, 470–471.
- Arbuj, S.S.; Waghmode, S.B.; Ramaswamy, A.V. Photochemical α-Bromination of Ketones Using N-Bromosuccinimide: A Simple, Mild and Efficient Method. Tetrahedron Lett. 2007, 48, 1411–1415.
- Wang, G.-W.; Gao, J. Solvent-Free Bromination Reactions with Sodium Bromide and Oxone Promoted by Mechanical Milling. Green Chem. 2012, 14, 1125–1131.
- Meshram, H.M.; Reddy, P.N.; Vishnu, P.; Sadashiv, K.; Yadav, J.S. A Green Approach for Efficient α-Halogenation of β-Dicarbonyl Compounds and Cyclic Ketones Using N-Halosuccinimides in Ionic Liquids. Tetrahedron Lett. 2006, 47, 991–995.
- Zou, L.-H.; Li, Y.-C.; Li, P.-G.; Zhou, J.; Wu, Z. Solvent-Controlled α-Monobromination, α,α-Dibromination or Imidation of 1,3-Diketones with N-Bromosuccinimide. Eur. J. Org. Chem. 2018, 2018, 5639–5643.
- Saikia, I.; Borah, A.J.; Phukan, P. Use of Bromine and Bromo-Organic Compounds in Organic Synthesis. Chem. Rev. 2016, 116, 6837–7042.
- Luo, L.; Meng, L.; Sun, Q.; Ge, Z.; Li, R. Novel Synthesis of Thiazolo/Thienoazepine-5,8-Diones from Dihalo Cyclic 1,3-Diketones and Mercaptonitrile Salts. RSC Adv. 2014, 4, 6845–6849.
- Mishra, A.K.; Nagarajaiah, H.; Moorthy, J.N. Trihaloisocyanuric Acids as Atom-Economic Reagents for Halogenation of Aromatics and Carbonyl Compounds in the Solid State by Ball Milling. Eur. J. Org. Chem. 2015, 2015, 2733–2738.
- Macharla, A.K.; Chozhiyath Nappunni, R.; Marri, M.R.; Peraka, S.; Nama, N. Oxidative Bromination of Ketones Using Ammonium Bromide and Oxone®. Tetrahedron Lett. 2012, 53, 191–195.
- Kim, J.-J.; Kweon, D.-H.; Cho, S.-D.; Kim, H.-K.; Lee, S.-G.; Yoon, Y.-J. Conversion of Nucleophilic Halides to Electrophilic Halides: Efficient and Selective Halogenation of Azinones, Amides, and Carbonyl Compounds Using Metal Halide/Lead Tetraacetate. Synlett 2006, 2006, 194–200.
- Košmrlj, J.; Kočevar, M.; Polanc, S. A New Convenient Bromination with KBrO3/KBr/Dowex®. Synth. Commun. 1996, 26, 3583–3592.
- Spitulnik, M.J. Synthesis of 1-Aryl-4-Halo-2-Pyrazolin-5-Ones by Ascorbic Acid Reduction of 1-Aryl-4,4-Dihalo-2-Pyrazolin-5-Ones. Synthesis 2002, 1985, 299–300.
- Stavber, G.; Zupan, M.; Jereb, M.; Stavber, S. Selective and Effective Fluorination of Organic Compounds in Water Using Selectfluor F-TEDA-BF4. Org. Lett. 2004, 6, 4973–4976.
- Matarlo, J.S.; Evans, C.E.; Sharma, I.; Lavaud, L.J.; Ngo, S.C.; Shek, R.; Rajashankar, K.R.; French, J.B.; Tan, D.S.; Tonge, P.J. Mechanism of MenE Inhibition by Acyl-Adenylate Analogues and Discovery of Novel Antibacterial Agents. Biochemistry 2015, 54, 6514–6524.
- Sloop, J.C.; Churley, M.; Guzman, A.; Moseley, S.; Stalker, S.; Weyand, J.; Yi, J. Synthesis and Reactivity of Fluorinated Cyclic Ketones: Initial Findings. Am. J. Org. Chem. 2014, 4, 1–10.
- Nyffeler, P.T.; Durón, S.G.; Burkart, M.D.; Vincent, S.P.; Wong, C.-H. Selectfluor: Mechanistic Insight and Applications. Angew. Chem. Int. Ed. 2005, 44, 192–212.
- Yi, H.; Zhang, G.; Wang, H.; Huang, Z.; Wang, J.; Singh, A.K.; Lei, A. Recent Advances in Radical C-H Activation/Radical Cross-Coupling. Chem. Rev. 2017, 117, 9016–9085.
- Shaw, M.H.; Twilton, J.; MacMillan, D.W.C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926.
- Wang, C.-S.; Dixneuf, P.H.; Soulé, J.-F. Photoredox Catalysis for Building C-C Bonds from C(Sp2)–H Bonds. Chem. Rev. 2018, 118, 7532–7585.
- Li, A.Y.; Moores, A. Carbonyl Reduction and Biomass: A Case Study of Sustainable Catalysis. ACS Sustain. Chem. Eng. 2019, 7, 10182–10197.
- Zhang, K.; Ma, R.; Wang, Y.; Shi, Z.; Lu, T.; Feng, J. Visible-Light-Promoted α,α-Dibromination in Minutes: Efficient Route for Construction of Quaternary Carbon Centers. ACS Sustain. Chem. Eng. 2019, 7, 18542–18546.
- Buckle, D.R.; Cantello, B.C.C.; Smith, H.; Spicer, B.A. 2-Cyano-1,3-Dicarbonyl Compounds with Antiallergic Activity. J. Med. Chem. 1977, 20, 265–269.
- Paquette, L.A.; Crich, D.; Fuchs, P.L.; Molander, G.A. Encyclopedia of Reagents for Organic Synthesis; Wiley: New York, NY, USA, 2009.
This entry is offline, you can click here to edit this entry!