Ploidy is the number of complete sets of chromosomes in a genome. Almost all vertebrate animals, including mammals, are diploid organisms whose somatic cells contain two sets of haploid genomes originally derived from each parent. However, a significant number of hepatocytes in the liver are physiologically polyploid, containing double, quadruple, or more chromosomes. Polyploidy of hepatocytes in the mammalian liver was originally suggested more than a century ago by the observation that the nuclear sizes of hepatocytes in adult rodent livers markedly increased compared with those in young livers [
1]. In addition, hepatocytes frequently contain more than one nucleus [
2]. These histological findings and a clear correlation between nuclear size and ploidy that was demonstrated later clarified polyploidization of hepatocytes during liver maturation [
1]. Furthermore, hepatocyte ploidy is known to increase during aging or liver damage. Although the mechanism underlying the regulation of the ploidy of hepatocytes and its effects on the pathophysiology of the liver are unclear, accumulating evidence indicates that polyploidy and alterations in the ploidy in hepatocytes influence liver homeostasis and diseases both positively and negatively. Whereas some reviews have outlined the advantages and disadvantages of polyploidy in health and disease in various organs [
3,
4], few papers have highlighted updated discussions about hepatocyte ploidy, especially ploidy alterations, and liver diseases [
5,
6,
7].
2. Physiological Polyploidization of Hepatocytes
2.1. Overview of Physiological Polyploidization of Hepatocytes
Physiological polyploidization of hepatocytes is widely observed in the liver of various mammalian species, including humans and rodents (
Figure 1A) [
1,
8]. Although the frequency of hepatocyte polyploidization differs depending on the species, physiological polyploidization is primarily observed during the course of liver maturation [
1,
8]. While almost all hepatocytes are diploid in neonatal mice and rats, polyploidization starts immediately prior to weaning in mice and at the time of weaning in rats [
9,
10,
11]. Most (~90%) hepatocytes in the adult rodent liver are polyploid [
12]. One of the triggers that induce drastic hepatocyte polyploidization in rodents is the transition in nutritional conditions associated with weaning, as discussed in the subsequent section [
10]. In contrast, the increase in physiological polyploidization in humans is slower than that in rodents. Approximately 95% of hepatocytes are diploid in newborns, and polyploid hepatocytes begin to appear in young children [
13]. The proportion of polyploid hepatocytes gradually increases with age and reaches up to 20–50% at an age of approximately 40–50 years [
13,
14,
15]. The ploidy levels of hepatocytes that increase during normal development are further enhanced with aging in both rodents and humans (
Figure 1B) [
13,
16]. Furthermore, intensified polyploidization in aged humans has been reported previously [
13].
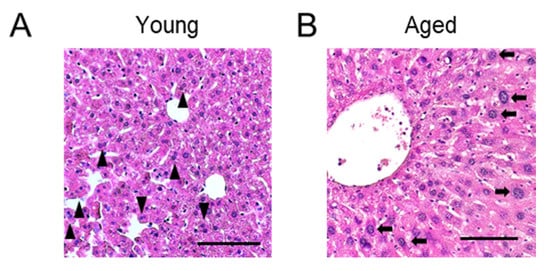
Figure 1. Histological images of livers in young (A) and aged (B) mice. Representative hematoxylin and eosin staining images are shown. (A) The liver of a 6-week-old mouse. Some binucleated polyploid hepatocytes are indicated by arrow heads. (B) The liver of a 69-week-old mouse. Polyploid hepatocytes with a large nucleus are indicated by arrows. Scale bars, 100 μm.
2.2. Mechanisms of Polyploidization
The mechanisms of polyploidization can be broadly classified into two types: abnormal cell cycle processes and cell fusion. In the former, genome duplication during the DNA synthesis (S) phase and subsequent abnormal cell cycle progression results in the formation of mononucleated or binucleated polyploid cells. In contrast, cell fusion can be induced in a cell cycle-independent manner and generates polyploid cells with two or more nuclei. Multinucleated skeletal muscle cells are formed by the fusion of mononucleated myoblasts during development [
17]. Syncytiotrophoblasts in placental villi are another example of polyploid cells produced by the cell fusion [
18].
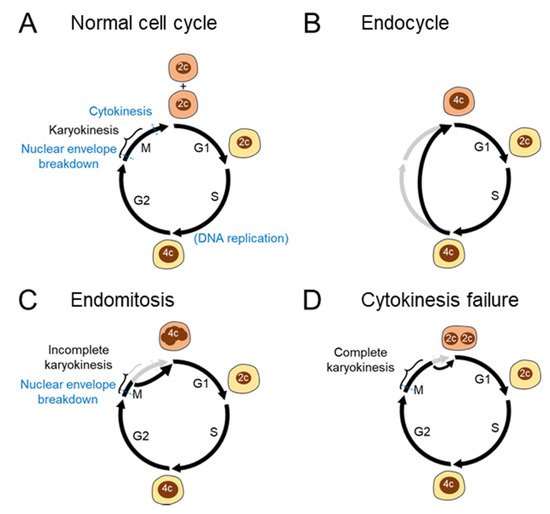
Polyploidization resulting from abnormal cell cycle processes can be classified in more detail (
Figure 2A–D). The endocycle is a variant cell cycle in which rounds of the S and gap (G) phases are repeated without entering the mitosis (M) phase (
Figure 2B). Owing to the lack of mitosis, endocyclic cells become polyploid without exhibiting the features of mitosis, including chromosome condensation and disruption of the nuclear envelope [
19]. Endocycles are relatively limited in mammals. Trophoblast giant cells are the best-studied cell types that undergo endocycles in mammals [
20]. Tubular epithelial cells in the kidney also undergo endocycles in response to acute kidney injury [
21].
In contrast to endocycles, endomitosis is a polyploidization process that is accompanied by mitosis (
Figure 2C). Megakaryocytes are a well-known polyploid cell type in mammals, and their polyploidization results from the abortion of mitosis during late anaphase [
22]. As chromosomes segregate incompletely during each M phase in polyploidizing megakaryocytes, polyploid megakaryocytes exhibit a polylobulated mononucleus [
22]. While such an endomitosis process without complete karyokinesis leads to the formation of mononucleated polyploid cells, failed cytokinesis after complete karyokinesis results in the formation of binucleated polyploid cells (
Figure 2D). Such an incomplete cytokinesis process is sometimes included as an extended definition of endomitosis. Cytokinesis failure is the primary mechanism underlying physiological polyploidization in hepatocytes and cardiomyocytes [
23]. Podocytes in the kidney are also unable to complete cytokinesis [
4], and multinucleated cells are physiologically observed in the kidney [
24]. Under diseased conditions, hepatocytes can become polyploid in other ways, including mitotic slippage and cell fusion, which will be discussed later.
Figure 2. Cell cycle aberrations leading to polyploidization (A) Normal cell cycle. A diploid cell gives rise to two diploid daughter cells through mitosis. (B) Endocycle. The S and G1 phases are repeated without entering the M phase, leading to the emergence of mononuclear polyploid cells. (C) Endomitosis. Although cells enter the M phase with chromosome condensation and disruption of the nuclear envelope, the mitotic process is aborted, leading to the genesis of mononucleated polyploid cells. (D) Cytokinesis failure. Cells complete karyokinesis but not the following cytokinesis, generating binucleated polyploid cells. 2c and 4c denote diploid and tetraploid DNA contents, respectively.
2.3. Molecular Mechanisms Underlying Physiological Polyploidization in Hepatocytes
The mechanism underlying the regulation of physiological polyploidization has not been completely elucidated. However, some key regulators have been demonstrated in rodent studies. The Desdouets group elegantly showed that after weaning, impaired organization of the actin cytoskeleton during late anaphase leads to the failure of RhoA activation required for cytokinesis in rat hepatocytes [
25]. In addition, insulin signaling, which can be enhanced by the suckling-to-weaning transition, induces cytokinesis failure in hepatocytes via activation of the downstream PI3K/Akt pathway in rats and mice [
10]. MicroRNAs, especially miR-122, are also important for inducing hepatocyte polyploidization [
11]. miR-122 is the predominant miRNA found in hepatocytes, and liver-specific miR-122 knockout reduces the number of binucleated hepatocytes by 60–70% in mice [
11]. Importantly, miR-122 is the most highly expressed miRNA in hepatocytes in 2–3-week-old mice when physiological binucleation of mouse hepatocytes is initiated, and miR-122 suppresses the expression of some cytokinesis-associated genes, including RhoA [
11]. Other genetically engineered mouse strains have also been reported to exhibit altered hepatocyte ploidy. Physiological polyploidization of hepatocytes is impaired in mice whose livers are deficient in atypical E2Fs, E2f7, and E2f8 [
26]. Between the two atypical E2F proteins, E2F8 seems to play a more important role in inducing hepatocyte polyploidization than E2F7, and E2f8-knockout and E2f7/E2f8 double-knockout livers predominantly comprise diploid hepatocytes [
26]. E2F8 is supposed to antagonize transcriptional activation by E2F1, and genes whose transcription is commonly targeted by both E2F1 and E2F8 include regulators of cytokinesis, such as Ect2, Mklp1, and Racgap [
26]. Genes associated with the circadian period are also involved in the physiological hepatocyte polyploidization [
27]. Hepatocyte polyploidization is markedly accelerated around the central vein in Per-null mice deficient in the three Period genes, namely, Per1, Per2, and Per3 [
27]. Mechanistically, Mkp1-mediated circadian modulation of the Erk1/2 activity is impaired in Per-null mice, which enhances the polyploidization [
27]. Moreover, other models in which regulators of cell cycles, such as RB, are manipulated also show altered ploidy status in hepatocytes [
28,
29]. Although it is still unclear whether these molecules play critical roles in the polyploidization of human hepatocytes, these findings provide a strong foundation for elucidating the detailed mechanisms underlying ploidy regulation in human hepatocytes.
2.4. Significance of Physiological Polyploidization
As physiological polyploidization of hepatocytes is a universal phenomenon in humans and rodents, physiologically polyploid hepatocytes would have some advantages in the development or maintenance of liver function. Typically, polyploidization is considered to induce terminal cellular differentiation. Physiological polyploidization in cardiomyocytes and megakaryocytes is accompanied by their differentiation [
30]. Hence, the physiological polyploidization observed during liver maturation may contribute to the enhancement of liver function.
To examine functional differences between diploid and polyploid hepatocytes, some studies have analyzed differentially expressed genes between them. However, gene expression seems to be almost unaffected by hepatocyte ploidy. Microarray and RNA sequencing analyses of sorted diploid and polyploid mouse hepatocytes did not identify clear transcriptional differences between diploid and polyploid hepatocytes [
31,
32]. On the other hand, some studies have suggested that polyploidy in hepatocytes affects cellular biological processes, such as lipid and carbohydrate metabolism; however, no consensus has been established with respect to the results [
33,
34,
35]. A recent study in which mouse liver cells were analyzed by single-nucleus RNA sequencing after ploidy-based sorting provided many important insights into the expression patterns of polyploid hepatocytes [
36]. In this study, Richter et al. showed that the transcriptomes of diploid and polyploid hepatocytes are globally very similar; however, some genes related to metabolism are differentially expressed between these two groups [
36]. Importantly, the expression of such ploidy-related genes is conditioned by their position within the hepatic lobule [
36]. In other words, polyploid hepatocytes are preferentially located in the pericentral zone, as previously suggested [
37,
38], and hepatic metabolic zone is the major determinant of gene expression levels in polyploid hepatocytes [
36]. In addition, impairment of hepatocyte polyploidization in E2f7/E2f8 double-knockout mice has no impact on liver differentiation, zonation, or metabolism [
26]. These findings challenge the traditional hypothesis that polyploidization promotes terminal differentiation in hepatocytes and is beneficial for adult liver function, including metabolism. The significance of polyploidy in liver function requires further study under both healthy conditions and those involving stress.
3. Polyploid Hepatocytes in Acute and Chronic Liver Damage
3.1. Ploidy Alterations in Liver Damage
Various types of liver injuries induce ploidy alterations in hepatocytes in the adult liver after physiological polyploidization. Enhancement of polyploidization has been observed in human chronic viral hepatitis [
15], as well as in nonalcoholic fatty liver disease [
41]. Some rodent models of liver injuries, such as those attributed to iron accumulation [
42], copper accumulation [
43], tyrosinemia [
44], and partial hepatectomy [
45], have also demonstrated enhanced hepatocyte polyploidization (
Figure 3A,B). Although it is difficult to discriminate between physiological polyploidization and a pathological one that may be induced by aging-related hepatocyte changes such as accumulation of DNA damages and dysfunctional mitochondria, polyploid hepatocytes also accumulate in aged livers [
6,
9].
Whereas physiological polyploidization of hepatocytes is predominantly induced by incomplete cytokinesis, polyploidization under pathological conditions can be attributed to various mechanisms. Gentric et al. showed that in livers with nonalcoholic fatty liver disease, DNA damage induced by oxidative stress leads to the inhibition of CDK1 activation and induces G2/M arrest via activation of the ATR/p53/p21 pathway, which results in polyploidization [
41]. A similar successive process that associates DNA damage and polyploidization has been reported during the induction of senescence [
47]. Various types of liver damage that accompany DNA damage and/or senescence induction in hepatocytes [
48] would lead to polyploidization of hepatocytes via arrest in the G2/M phase mediated by p53 activation. Mitotic slippage observed after prolonged spindle assembly checkpoint (SAC) activation is another type of abnormal cell cycle progression that leads to polyploidization [
49]. SAC ensures accurate attachment between chromosomes and spindle microtubules, and its activation prevents the progression from metaphase to anaphase. Inextricable SAC activation induces mitotic cell death but is sometimes bypassed, leading to premature exit from mitosis, which is called mitotic slippage [
49]. Mitotic slippage is particularly observed in cancer cells treated with microtubule-affecting drugs, and it generates mononucleated polyploid cells [
49]. In addition, the hepatitis B virus (HBV) X protein is reported to bind to BUB1B, a component of the mitotic checkpoint complex, and enhance the mitotic slippage [
50]. HBV X protein also induces abortive mitosis and abnormal polyploidization by activating p38 and upregulating the expression of PLK1 [
51]. Consistently, human hepatocytes infected with HBV show an enhanced polyploidization [
51]. Moreover, cell fusion can be observed in an injured liver. Cell fusion between hepatocytes and bone marrow-derived macrophages is observed when injured livers of fumarylacetoacetate hydrolase (Fah)-deficient mice, a model of familial tyrosinemia type I, are regenerated by the transplantation of bone marrow cells [
52,
53].
Interestingly, both polyploidization and reduction of ploidy occur in injured livers. The generation of diploid hepatocytes from polyploid cells was first demonstrated in the fusion-derived hepatocytes mentioned above [
54]. Moreover, the transplantation of sorted polyploid hepatocytes into the recipient livers resulted in the emergence of diploid cells during the proliferation of donor polyploid cells [
55]. Such ploidy reduction (depolyploidization) occurs via multipolar mitosis [
55] and is detected during various chronic liver injuries [
37]. Intriguingly, depolyploidized diploid cells readily undergo re-polyploidization in subsequent cell cycles [
37,
55]. Thus, ploidies of hepatocytes can be altered dynamically during liver injury, and polyploidization and depolyploidization are induced by various mechanisms under pathological conditions.
3.2. Liver Regeneration by Polyploid Hepatocytes
As described previously, polyploidization is considered to be associated with terminal cell differentiation, and the proliferation of terminally differentiated polyploid cells is arrested in many cell types [
19,
30]. In some organ systems, development and regeneration stem exclusively from diploid cells, whereas polyploid cells function as differentiated cells and do not proliferate for regeneration. For example, hematopoietic stem/progenitor cells are diploid, and differentiated polyploid megakaryocytes do not proliferate [
22]. Heart regeneration is also attributed to diploid cardiomyocytes, although there is lack of clarity regarding whether polyploid cardiomyocytes are completely unable to proliferate [
56,
57]. These observations, as well as the fact that polyploidization frequently induces cell cycle arrest in nontransformed cells in vitro [
58], led to the hypothesis of the “tetraploidy checkpoint” in the past [
59].
However, hepatocytes do not seem to have such a definitive “polyploidy checkpoint,” and they retain their proliferative capacities even after polyploidization. The liver has an extensive regenerative capacity, and the proliferation of hepatocytes primarily leads to the regeneration of the liver damaged by surgical resection or various liver injuries. After 70% partial hepatectomy in mice, during which the most dynamic liver regeneration is observed, almost all hepatocytes, including polyploid cells, enter cell cycle and undergo DNA synthesis in the S phase [
60]. Approximately half of these hepatocytes do not enter the M phase, resulting in polyploidization and hepatocyte hypertrophy, whereas others complete mitosis and proliferate [
60]. Polyploid hepatocytes proliferate in this way after massive surgical resection of the liver, although the initiation of the proliferation of polyploid hepatocytes for regeneration after hepatectomy is reported to be relatively delayed compared to that of diploid hepatocytes [
61].
In contrast to such transient liver regeneration, whether polyploid hepatocytes continuously proliferate in chronic liver injuries remained unclear because it was difficult to distinguish between the emergence of polyploid cells by polyploidization of diploid cells and polyploid cell proliferation. A novel system to trace polyploidy using multicolor reporter mice was used to overcome this challenge, and the results confirmed the persistent proliferation of polyploid hepatocytes in chronically damaged livers [
37]. In mice heterozygous for a multicolor reporter allele, polyploid cells are labeled by co-expression of multiple colors, and the proliferation of polyploid cells can be traced [
37]. This system clearly demonstrated that the majority of polyploid hepatocytes, even octaploid hepatocytes, proliferate continuously during various types of chronic liver injury [
37]. Notably, proliferating multicolored polyploid hepatocytes sometimes give rise to monocolored daughter cells, indicating ploidy reduction during regeneration driven by polyploid cells in chronically injured livers [
37]. Moreover, polyploid hepatocytes repeatedly divide to maintain normal hepatocyte turnover during aging [
16]. Taken together, these findings indicate that abundant polyploid hepatocytes have extensive regenerative potential and participate in liver regeneration.