Here we talk about the effects of acidosis on insulin signaling and glucose uptake in skeletal muscle and whether correcting defects that maintain [pH]i within the muscle, such as carnosine, could alleviate insulin resistance improve insulin responses during metabolic syndrome.
- Skeletal muscle
- Insulin signaling
- Glycolysis
- Intracellular acidosis
- Carnosine
1. Introduction
Diabetes and obesity-related diseases afflict 425 million people worldwide and have emerged as a significant cause of mortality and morbidity in both developed and developing countries. Type 2 diabetes (T2D) is associated with a decrease in number and defects in the function of insulin-producing pancreatic β-cells. However, the decreased response of skeletal muscle to insulin is evident even before pancreatic β-cell failure [1][2]. Skeletal muscle is the principal site for insulin-stimulated glucose uptake, which consumes approximately 70–80% of the glucose via insulin-dependent mechanisms [3]. Hence, defects in insulin-stimulated muscle glucose uptake are considered a principal component of typical obesity-associated insulin resistance [4][5]. Glucose homeostasis in the muscle is a complex interplay between insulin signaling and glucose utilization. However, a consistent finding is that maintaining proper insulin responses of the IRS1–PI3K–AKT signaling pathway within the skeletal muscle is central to maintaining glucose homeostasis. This pathway is markedly impaired in the muscle of humans with type 2 diabetes (T2D), healthy glucose-tolerant offspring of parents with T2D, obese non-diabetics, people with non-alcoholic fatty liver disease, and chronic kidney disease (CKD) [6][7][8][9][10][11][12]. Defects in the insulin-signaling pathway are traceable to defective insulin receptor (IR) kinase activity, a shift from tyrosine to serine phosphorylation in IRS1, and blunted IRS1-associated P13K activity, followed by a subsequent decrease in AKT phosphorylation [7]. Much attention in the field has been focused on the defects of insulin-mediated metabolic actions via the IRS1–PI3K–AKT signaling pathway in the muscle. However, in addition to defects in this signaling pathway, numerous reports suggest that other pathways, such as inflammation [13][14][15][16], lipid peroxidation products [17], oxidative stress [18][19][20][21][22][23], a decrease in intracellular pH [pH]i [9][24] and autophagy [25][26], could cross-talk with insulin-signaling pathway and either cause or exacerbate insulin resistance in the muscle. In this article, we will highlight a few issues, which are central to the gap in knowledge about whether the defects in glycolysis, within the muscle, are causal or secondary to insulin resistance. We will discuss, whether the increase in the interconversion of pyruvate to lactic acid, which subsequently dissociates to lactate and hydrogen (H+) ions, in the skeletal muscle of T2D humans, could contribute to a decrease in [pH]i. We discuss whether the defects in H+ ion transporters and depletion of intracellular histidyl dipeptides, in the skeletal muscle, could contribute towards a decrease in [pH]i and be a causative factor of insulin resistance. Furthermore, we will highlight studies suggesting the effect of a decrease in [pH]i on the intermediates of the glycolytic pathway and protein degradation. Finally, we will highlight, whether correcting defects in pathways which maintain [pH]i within the muscle, could alleviate insulin resistance and improve insulin responses during metabolic syndrome.
2. Maintenance of Intracellular pH ([pH]i) in the Muscle
Regulation of [pH]i within the muscle is a complex process, which is maintained by several membrane transporters and endogenous buffers [27]. In the muscle, pH homeostasis is maintained by a complex transporter system that involves the Na+/H+ exchanger (NHE), Na+-dependent and independent bicarbonate systems (NBC) and monocarboxylate transporters (MCT1 and 4) [27][28][29][30][31]. In the skeletal muscle, the NHE system is considered the most important regulatory system, which is active at resting levels. However, a minor fraction of H+ ions are released by the NHE during exercise [32]. In rats fed with low doses of streptozotocin and a high-fat diet, NHE expression was decreased in the soleus and extensor digitorum longus muscles, which was increased by endurance training [33]. There are at least two isoforms of the NBC transport systems, NBCe1 and NBCe2, present in the human skeletal muscle; however, the contribution of these transporters to pH regulation is not clear [34]. Studies with diabetic rats showed that NBC expression remains unchanged and endurance training increases NBC expression in the muscle [33].
In addition to the NHE and NBC, two monocarboxylate transporters, MCT1 and MCT4, present in the muscle predominantly facilitate H+ ions and lactate efflux (symporter) across the plasma membrane during exercise [29]. MCT1 is ubiquitously expressed, whereas MCT4 is mainly expressed in the glycolytic muscle fibers. Skeletal muscle is made up of different bundles of muscle fibers, which are broadly classified as slow-twitch (Type 1) and fast-twitch (Type 2) fibers. Based on the myosin heavy chain gene expression (MYH), fast-twitch is further classified as Type 2a, 2X, and 2B. The skeletal muscle fibers vary in energy production, with Type 1 and 2A primarily using the oxidative metabolism and type 2X and 2B fibers using the glycolytic metabolism [35][36]. Numerous studies show that MCT1 and MCT4 protein expression is increased in the skeletal muscle after exercise training and electrical stimulation in healthy humans [37][38][39]. MCT1 expression is increased after endurance training and its expression is correlated with the oxidative capacity [40], whereas MCT4 expression is increased following intensive exercise and related to the indexes of glycolytic metabolism [41]. Because MCT1 is mostly found in oxidative fibers and displays a higher affinity for L-lactate compared with MCT4, it has been suggested that MCT1 mainly handles the uptake of lactate and H+ ions, whereas MCT4 is involved in lactate and H+ efflux [42][43][44][45]. However, all the MCTs can operate in both directions, involving the efflux and influx of lactate and H+ ions. In humans with T2D, MCT1 expression is decreased in the skeletal muscle compared with healthy humans [46]. Studies with heterozygous MCT1+/− mice show that the [pH]i within the muscle at rest was higher compared with the wild type mice, but the drop in [pH]i was higher in the MCT1+/− mice during the initial minutes of exercise. Hence, MCTI is involved in the homeostatic control of pH within the skeletal muscle both at rest and during exercise [28]. MCT1+/− mice displayed normal insulin sensitivity, whereas MCT1+/− mice fed with a high-fat diet were resistant to diet-induced obesity, insulin resistance, and glucose intolerance. This phenotype was associated with reduced food intake and decreased intestinal absorption under high-fat diet conditions [47]. Given that MCT1 handles H+ influx [42][43][44][45], it could be speculated that MCT1 deletion, diminishes the uptake of H+ ions into the muscle, maintains alkaline pH, and thus preserves the insulin responses during high-fat feeding. In addition to these transport systems, the skeletal muscle contains at least four isozymes of carbonic anhydrase (CA II, III, IV and V) that accelerate the removal of acid as CO2 [48]. Proteomic profiling of crude muscle extracts from the non-obese Goto–Kakizaki rat model of T2D identified that the expression of CA III had the highest decrease compared with the non-obese mice [49]. Since the CA III isoform is one of the faster enzymes which helps regulate [pH]i, the decrease in its expression could also decrease the removal of acid in diabetic muscle. Indeed studies with CA III knockout mice showed that the drop in [pH]i following intense stimulation of the gastrocnemius muscle was significantly more in the CA III knockout mice compared with the wild type mice [50].
Taken together, these studies suggest that the essential components of the pH regulatory systems are imbalanced in T2D muscle. Further, the improvement in insulin action by endurance training could be facilitated by increased expression of both NHE and MCT1 transporters in the skeletal muscle [51][52][53][54][55], which could enhance the removal of H+ ions and thus improve buffering capacity.
3. Histidyl Dipeptides and Intracellular pH [pH]i Regulation
Histidyl dipeptides have a pKa value close to the physiological pH (6.8–7.1), compared with the bicarbonate (pKa 6.3), inorganic phosphate (pKa 7.2), and histidine (pKa 6.2) [56], which renders them ideally suited to act as intracellular buffers within the pH transit range of the skeletal muscle. Theoretical estimates predict that these dipeptides could contribute to approximately 7–40% of muscle buffering capacity [57][58]. In comparison with high molecular weight transporters and enzymes, which possess lower intracellular mobility and restrict their capacity to buffer protons and correct the [pH]i, histidyl dipeptides are small molecular weight dipeptides with a molecular weight range of 227–241 Da. These dipeptides diffuse reversibly two or more orders of magnitude faster than proteins across the cell [59]. Skeletal muscle is the largest reservoir of histidyl dipeptides, and approximately 10–20 mM levels of these dipeptides are present in the muscle [59][60]. Among the naturally occurring histidyl dipeptides, carnosine is the most common dipeptide present in human skeletal muscle, whereas its methylated analogs, anserine (β-alanine-Nπ-histidine) and balenine (β-alanine-Nτ-histidine) are largely found in other mammalian and avian species [59][60][61].
Carnosine is synthesized via the ATP grasp protein carnosine synthase (Carns1), which ligates the non-proteinogenic amino acid β-alanine with histidine to form carnosine and further methylates to anserine by carnosine methyltransferase [62][63][64][65]. In addition to [pH]i buffering [59][66], these dipeptides also scavenge lipid peroxidation products, such as 4-hydroxy trans-2-nonenal (HNE) [67][68], quench reactive oxygen species (ROS) such as singlet oxygen [69], and chelate first transition metals [70]. Recent reports from our laboratory and others showed that carnosine levels are increased upon exercise and significantly depleted in the muscle of normal humans and T2D patients, respectively [60][71]. We also tested whether the levels of histidyl dipeptides are affected in the skeletal muscle by high-fat feeding in rodent models. For these studies, we performed a pilot study and fed the wild type C57 mice (7–8 weeks) with a high-fat and high-sucrose (HFHS) diet for 12–14 weeks. Gastrocnemius muscle from the normal chow (NC)- and HFHS-fed mice were analyzed by LC-MS for carnosine and anserine levels. In parallel with the observations in T2D humans that histidyl dipeptides are depleted in the gastrocnemius muscle [71], we found that carnosine levels in the gastrocnemius muscles of HFHS-fed mice were significantly decreased compared with the NC-fed mice (Figure 1). Taken together, these reports showing that carnosine levels change under divergent conditions suggest that histidyl dipeptides may be essential for regulating specific metabolic processes such as [pH]i under physiological and pathological conditions.
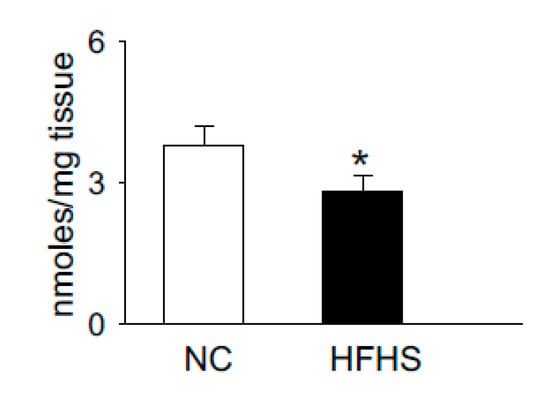
Figure 1. High-fat high-sucrose (HFHS) feeding to wild type (WT) mice decreases histidyl dipeptides in skeletal muscle. WT C57Bl6 mice (8 weeks old) were fed with normal chow (NC) and high-fat high-sucrose feeding for 12 weeks. Gastrocnemius muscles were isolated from the mice after NC and HFHS feeding, which was analyzed by LC-MS for anserine and carnosine levels, using tyrosine histidine as an internal standard. Data are presented as mean ± SEM, n = 5–6 mice in each group, * p < 0.05 vs NC.
It has been reported that carnosine supplementation in different animal models of metabolic syndrome could alleviate some markers of metabolic syndrome. Studies with high fat-fed mice supplemented with histidine, β-alanine, or carnosine showed that histidine and carnosine supplementation reduced the activities of lipogenic enzymes and sterol regulatory element-binding proteins in the liver. These changes were also accompanied by improved insulin sensitivity and hyperinsulinemia [72]. Carnosine feeding to obese Zucker rats enhanced the extrusion of lipid peroxidation products, such as 4HNE, by forming carnosine aldehyde conjugates, diminishing carbonyl stress, dyslipidemia, and hypertension [67]. Similarly, carnosine supplementation to db/db mice increased the pancreatic islet sizes and insulin release [73]. In parallel with the reports from animal models of diabetes, recent studies with obese insulin-resistant humans showed that carnosine supplementation modestly attenuates insulin release and enhances the extrusion of reactive aldehydes in urine [74][75]. However, a major obstacle for clinical applicability with these dipeptides is that they are hydrolyzed by carnosinase (CNDP1) present in the serum, which diminishes their bioavailability at the site of injury [76]. To circumvent this challenge, several groups tested the carnosine analogs, which are resistant to hydrolysis, such as D-carnosine. However, these carnosinase-resistant enantiomers have poor absorption and failed in testing [77]. Similarly, the octyl-D ester derivative of D-carnosine, which had better absorption issues, failed due to dosing limitations [78]. Recent findings show supplementation of another carnosine analog, carnosinol, alleviates carbonyl stress, and diminishes some markers of diet-induced obesity in the rodent model. Carnosinol supplementation decreased HNE adduct formation in the liver and skeletal muscle, mitigated inflammation, insulin resistance, and steatohepatitis [17].
Homeostasis of histidyl dipeptides within the muscle is regulated by a complex machinery of transporters (PEPT 1 and 2, TAUT), Carns1 activity, lipid peroxidation products, and carnosinase 1 and 2 (CNDP), which hydrolyze carnosine to β-alanine and histidine [62][66][79]. It is not clear how these dipeptides are depleted in the muscle during metabolic syndrome. Previous studies with humans showed that the generation of lipid peroxidation products is increased, and buffering capacity is diminished in the skeletal muscle of T2D subjects [80][81]. Henceforth, it is possible that histidyl dipeptides act as sacrificial peptides that are used by excessive generation of reactive aldehydes or consumed to buffer the changes in [pH]i. In addition, it can be speculated that the decrease in Carns1 activity might also contribute to a decrease in carnosine levels.
Despite the suggestive and circumstantial evidence showing the beneficial effects of carnosine and its analogs on metabolic syndrome, it is not clear whether the depletion of carnosine in the muscle contributes to decreasing buffering capacity and is causative of insulin resistance [80]. Similarly, it is not clear whether the depletion of histidyl dipeptides affects protein folding and autophagic response within T2D muscle. Additionally, it is not clear, whether the supplementation of carnosine or its analogs to obese mice models or humans replenishes histidyl dipeptides and improves buffering capacity and autophagic responses within the muscle. Moreover, the role of enzyme Carns1, which synthesizes these dipeptides [62], has never been properly studied. Hence, a comprehensive understanding of how histidyl dipeptides regulate muscle function under physiological and pathological conditions could be elucidated by generating skeletal muscle-specific Carns-null or transgenic mice. Generation of Carns-null mice models will help to understand whether depleting histidyl dipeptides within the muscle affects skeletal muscle pH, insulin signaling, and autophagy. Further, Carns overexpression in the skeletal muscle will specifically elucidate whether replenishing these dipeptides in muscle during metabolic syndrome could alleviate buffering potential and prevent protein damage and insulin resistance. Furthermore, these mice models will help understand which biochemical property, such as [pH]i buffering or aldehyde quenching by these dipeptides, is essential to preserve skeletal muscle function under pathological and physiological conditions. Elucidation of these pathways will help synthesize analogs that could specifically enhance the buffering or aldehyde quenching capacity in the skeletal muscle and affect the insulin responses during metabolic syndrome.
4. Conclusions
Skeletal muscle comprises a large percentage of the body’s mass, handles about 70–80% of the insulin-stimulated glucose, and contributes to approximately 25% of lactate formation. Derangements in glucose and fatty acid utilization within the muscle is considered to be the underlying cause of insulin resistance. In the insulin-resistant state, there is a decrease in both the uptake of glucose and key regulators of the glycolytic pathway. Studies with genetically modified mice models indicated that the overexpression or deletion of key glycolytic enzymes had either minimal effect on the obese metabolic phenotype or caused insulin resistance [82][83]. However, to fully understand, whether the defects in the glycolytic pathway contributes to insulin resistance, tracer studies using 13C-labelled glucose are needed to understand and determine which step in the glycolytic pathway is affected and whether correction of that step could preserve insulin signaling during metabolic syndrome.
Within the skeletal muscle of T2D patients, there is increased interconversion of pyruvate to lactate [84]. Since lactate is released from the dissociation of lactic acid to lactate and H+ ions, increased pyruvate to lactate interconversion could drop the [pH]i within the skeletal muscle of T2D patients. Additionally, a decrease in the expression of H+ ion transporters and histidyl dipeptide levels could have a systemic effect on the [pH]i and contribute to diminishing the buffering capacity within the skeletal muscle of T2D humans [80] (Figure 2). Although in vitro and in vivo studies indicate that the change in [pH]i causes insulin resistance, the mechanisms by which drop in [pH]i contributes to decreasing insulin responses are not clearly defined. Given that skeletal muscle is the main target of insulin resistance, hence it is essential to develop a comprehensive understanding of the mechanisms, which imbalance [pH]i and contribute to diminishing insulin responses. An understanding of these readouts could help develop etiology-based countermeasures to alleviate insulin responses in the muscle during insulin-resistant states, such as T2D and CKD.
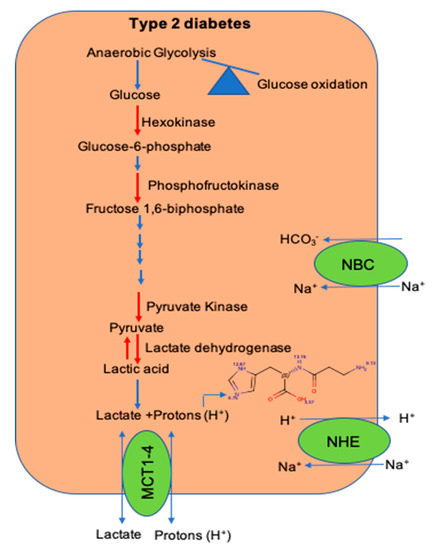
Figure 2. The shift in glucose utilization and imbalance of pathways that maintain intracellular pH during diet-induced metabolic syndrome. A shift in glucose utilization from glucose oxidation to anaerobic glycolysis enhances lactate production and hydrogen (H+) ions. Within the skeletal muscle, the activity of key glycolytic enzymes hexokinase and pyruvate kinase is decreased during metabolic syndrome. Expression of transporters; the Na-H+ (NHE) exchanger, which transports hydrogen (H+) ions during exercise and rest, respectively; and levels of histidyl dipeptides, which buffer H+ ions, are decreased during diet-induced metabolic syndrome.
This entry is adapted from the peer-reviewed paper 10.3390/nu12102910
References
- Lillioja, S.; Mott, D.M.; Howard, B.V.; Bennett, P.H.; Yki-Jarvinen, H.; Freymond, D.; Nyomba, B.L.; Zurlo, F.; Swinburn, B.; Bogardus, C. Impaired Glucose Tolerance as a Disorder of Insulin Action. Longitudinal and cross-sectional studies in Pima Indians. N. Eng. J. Med. 1988, 318, 1217–1225.
- Warram, J.H.; Martin, B.C.; Krolewski, A.S.; Soeldner, J.S.; Kahn, C.R. Slow Glucose Removal Rate and Hyperinsulinemia Precede the Development of Type II Diabetes in the Offspring of Diabetic Parents. Ann. Intern. Med. 1990, 113, 909–915.
- Stump, C.S.; Henriksen, E.J.; Wei, Y.; Sowers, J.R. The metabolic syndrome: Role of skeletal muscle metabolism. Ann. Med. 2006, 38, 389–402.
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163.
- Shulman, G.I. Ectopic Fat in Insulin Resistance, Dyslipidemia, and Cardiometabolic Disease. N. Eng. J. Med. 2014, 371, 1131–1141.
- Krook, A.; Björnholm, M.; Galuska, D.; Jiang, X.J.; Fahlman, R.; Myers, M.G.; Wallberg-Henriksson, H.; Zierath, J.R. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes 2000, 49, 284–292.
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223.
- Bouzakri, K.; Koistinen, H.; Zierath, J.R. Molecular Mechanisms of Skeletal Muscle Insulin Resistance in Type 2 Diabetes. Curr. Diabetes Rev. 2005, 1, 167–174.
- Bailey, J.L.; Zheng, B.; Hu, Z.; Price, S.R.; Mitch, W.E. Chronic Kidney Disease Causes Defects in Signaling through the Insulin Receptor Substrate/Phosphatidylinositol 3-Kinase/Akt Pathway: Implications for Muscle Atrophy. J. Am. Soc. Nephrol. 2006, 17, 1388–1394.
- Deivanayagam, S.; Mohammed, B.S.; E Vitola, B.; Naguib, G.H.; Keshen, T.H.; Kirk, E.P.; Klein, S. Nonalcoholic fatty liver disease is associated with hepatic and skeletal muscle insulin resistance in overweight adolescents. Am. J. Clin. Nutr. 2008, 88, 257–262.
- Kobayashi, S.; Maesato, K.; Moriya, H.; Ohtake, T.; Ikeda, T. Insulin resistance in patients with chronic kidney disease. Am. J. Kidney Dis. 2005, 45, 275–280.
- Nerpin, E.; Risérus, U.; Ingelsson, E.; Sundström, J.; Jobs, M.; Larsson, A.; Basu, S.; Ärnlöv, J. Insulin Sensitivity Measured With Euglycemic Clamp Is Independently Associated With Glomerular Filtration Rate in a Community-Based Cohort. Diabetes Care 2008, 31, 1550–1555.
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119.
- Perreault, M.; Marette, A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat. Med. 2001, 7, 1138–1143.
- Pedersen, B.K.; Steensberg, A.; Schjerling, P. Muscle-derived interleukin-6: Possible biological effects. J. Physiol. 2001, 536, 329–337.
- Bruunsgaard, H. Physical activity and modulation of systemic low-level inflammation. J. Leukoc. Biol. 2005, 78, 819–835.
- Anderson, E.J.; Vistoli, G.; Katunga, L.A.; Funai, K.; Regazzoni, L.; Monroe, T.B.; Gilardoni, E.; Cannizzaro, L.; Colzani, M.; De Maddis, D.; et al. A carnosine analog mitigates metabolic disorders of obesity by reducing carbonyl stress. J. Clin. Investig. 2018, 128, 5280–5293.
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761.
- Urakawa, H.; Katsuki, A.; Sumida, Y.; Gabazza, E.C.; Murashima, S.; Morioka, K.; Maruyama, N.; Kitagawa, N.; Tanaka, T.; Hori, Y.; et al. Oxidative Stress Is Associated with Adiposity and Insulin Resistance in Men. J. Clin. Endocrinol. Metab. 2003, 88, 4673–4676.
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948.
- Maddux, B.A.; See, W.; Lawrence, J.C., Jr.; Goldfine, A.L.; Goldfine, I.D.; Evans, J.L. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by mircomolar concentrations of alpha-lipoic acid. Diabetes 2001, 50, 404–410.
- Rudich, A.; Tirosh, A.; Potashnik, R.; Hemi, R.; Kanety, H.; Bashan, N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 1998, 47, 1562–1569.
- Rudich, A.; Tirosh, A.; Potashnik, R.; Khamaisi, M.; Bashan, N. Lipoic acid protects against oxidative stress induced impairment in insulin stimulation of protein kinase B and glucose transport in 3T3-L1 adipocytes. Diabetologia 1999, 42, 949–957.
- Franch, H.A.; Raissi, S.; Wang, X.; Zheng, B.; Bailey, J.L.; Price, S.R. Acidosis impairs insulin receptor substrate-1-associated phosphoinositide 3-kinase signaling in muscle cells: Consequences on proteolysis. Am. J. Physiol. Physiol. 2004, 287, F700–F706.
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515.
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.-H.; Cho, J.M.; Kim, Y.-N.; Kim, S.S.; Kim, H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2012, 19, 83–92.
- Juel, C. Regulation of pH in human skeletal muscle: Adaptations to physical activity. Acta Physiol. Oxf. 2008, 193, 17–24.
- Chatel, B.; Bendahan, D.; Hourdé, C.; Pellerin, L.; Lengacher, S.; Magistretti, P.J.; Le Fur, Y.; Vilmen, C.; Bernard, M.; Messonnier, L.A. Role of MCT1 and CAII in skeletal muscle pH homeostasis, energetics, and function: In vivo insights from MCT1 haploinsufficient mice. FASEB J. 2017, 31, 2562–2575.
- Halestrap, A.P.; Price, N.T. The proton-linked monocarboxylate transporter (MCT) family: Structure, function and regulation. Biochem. J. 1999, 343, 281–299.
- Halestrap, A.P.; Meredith, D. The SLC16 gene family?from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflügers Arch. 2004, 447, 619–628.
- Meldrum, N.U.; Roughton, F.J.W. Carbonic anhydrase. Its preparation and properties. J. Physiol. 1933, 80, 113–142.
- Juel, C. Muscle pH regulation: Role of training. Acta Physiol. Scand. 1998, 162, 359–366.
- Monazzami, A.; Rajabi, H.; Ghrakhanlou, R.; Yari, K.; Rahimi, Z. Modulation of oxidative and glycolytic skeletal muscle fibers Na+/H+ exchanger1 (NHE1) and Na+/HCO3- co-transporter1 (NBC1) genes and proteins expression in type 2 diabetic rat (Streptozotocin + high fat diet) following long term endurance training. Cell Mol. Biol. Noisy Grand 2017, 63, 11–18.
- Kristensen, J.M.; Kristensen, M.; Juel, C. Expression of Na+/HCO3- co-transporter proteins (NBCs) in rat and human skeletal muscle. Acta Physiol. Scand. 2004, 182, 69–76.
- Schiaffino, S.; Reggiani, C. Fiber Types in Mammalian Skeletal Muscles. Physiol. Rev. 2011, 91, 1447–1531.
- Pette, D.; Staron, R.S. Myosin isoforms, muscle fiber types, and transitions. Microsc. Res. Tech. 2000, 50, 500–509.
- Bonen, A.; Mccullagh, K.J.A.; Putman, C.T.; Hultman, E.; Jones, N.L.; Heigenhauser, G.J.F. Short-term training increases human muscle MCT1 and femoral venous lactate in relation to muscle lactate. Am. J. Physiol. Content 1998, 274, E102–E107.
- Pilegaard, H.; Mohr, T.; Kjaer, M.; Juel, C. Lactate/H+ transport in skeletal muscle from spinal-cord-injured patients. Scand. J. Med. Sci. Sports 1998, 8, 98–101.
- Dubouchaud, H.; Granier, P.; Mercier, J.; Le Peuch, C.; Préfaut, C. Lactate uptake by skeletal muscle sarcolemmal vesicles decreases after 4 wk of hindlimb unweighting in rats. J. Appl. Physiol. 1996, 80, 416–421.
- Dubouchaud, H.; Butterfield, G.E.; Wolfel, E.E.; Bergman, B.C.; Brooks, G.A. Endurance training, expression, and physiology of LDH, MCT1, and MCT4 in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E571–E579.
- Bonen, A. Lactate transporters (MCT proteins) in heart and skeletal muscles. Med. Sci. Sports Exerc. 2000, 32, 778–789.
- Juel, C.; Halestrap, A.P. Lactate transport in skeletal muscle - role and regulation of the monocarboxylate transporter. J. Physiol. 1999, 517, 633–642.
- Pilegaard, H.; Domino, K.; Noland, T.; Juel, C.; Hellsten, Y.; Halestrap, A.P.; Bangsbo, J. Effect of high-intensity exercise training on lactate/H+ transport capacity in human skeletal muscle. Am. J. Physiol. Content 1999, 276, E255–E261.
- Pilegaard, H.; Terzis, G.; Halestrap, A.; Juel, C. Distribution of the lactate/H+ transporter isoforms MCT1 and MCT4 in human skeletal muscle. Am. J. Physiol. Content 1999, 276, E843–E848.
- Wilson, M.C.; Jackson, V.N.; Heddle, C.; Price, N.T.; Pilegaard, H.; Juel, C.; Bonen, A.; Montgomery, I.; Hutter, O.F.; Halestrap, A.P. Lactic Acid Efflux from White Skeletal Muscle Is Catalyzed by the Monocarboxylate Transporter Isoform MCT3. J. Biol. Chem. 1998, 273, 15920–15926.
- Juel, C.; Holten, M.K.; Dela, F. Effects of strength training on muscle lactate release and MCT1 and MCT4 content in healthy and type 2 diabetic humans. J. Physiol. 2004, 556, 297–304.
- Lengacher, S.; Nehiri-Sitayeb, T.; Steiner, N.; Carneiro, L.; Favrod, C.; Preitner, F.; Thorens, B.; Stehle, J.-C.; Dix, L.; Pralong, F.; et al. Resistance to Diet-Induced Obesity and Associated Metabolic Perturbations in Haploinsufficient Monocarboxylate Transporter 1 Mice. PLoS ONE 2013, 8, e82505.
- Wetzel, P.; Gros, G. Carbonic Anhydrases in Striated Muscle. In Experientia Supplementum; The Carbonic Anhydrases: New Horizons; Chegwidden, W.R., Carter, N.D., Edwards, Y.H., Eds.; Birkhäuser: Basel, Switzerland; pp. 375–399.
- Mullen, E.; Ohlendieck, K. Proteomic profiling of non-obese type 2 diabetic skeletal muscle. Int. J. Mol. Med. 2010, 25, 445–458.
- Liu, M.; Walter, G.A.; Pathare, N.C.; Forster, R.E.; Vandenborne, K. A quantitative study of bioenergetics in skeletal muscle lacking carbonic anhydrase III using 31P magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. USA 2006, 104, 371–376.
- Christ, C.Y.; Hunt, D.; Hancock, J.; García-Macedo, R.; Mandarino, L.J.; Ivy, J.L. Exercise training improves muscle insulin resistance but not insulin receptor signaling in obese Zucker rats. J. Appl. Physiol. 1985 2002, 92, 736–744.
- Hevener, A.L.; Reichart, D.; Olefsky, J. Exercise and thiazolidinedione therapy normalize insulin action in the obese Zucker fatty rat. Diabetes 2000, 49, 2154–2159.
- Becker-Zimmermann, K.; Berger, M.; Berchtold, P.; Gries, F.A.; Herberg, L.; Schwenen, M. Treadmill training improves intravenous glucose tolerance and insulin sensitivity in fatty zucker rats. Diabetologia 1982, 22, 468–474.
- Metz, L.; Vermaelen, M.; Lambert-Cordillac, K.; Broca, C.; Sirvent, P.; Raynaud, E.; Mercier, J. Endurance training increases lactate transport in male Zucker fa/fa rats. Biochem. Biophys. Res. Commun. 2005, 331, 1338–1345.
- Nikooie, R.; Rajabi, H.; Gharakhanlu, R.; Atabi, F.; Omidfar, K.; Aveseh, M.; Larijani, B. Exercise-induced changes of MCT1 in cardiac and skeletal muscles of diabetic rats induced by high-fat diet and STZ. J. Physiol. Biochem. 2013, 69, 865–877.
- Abe, H. Role of histidine-related compounds as intracellular proton buffering constituents in vertebrate muscle. Biochem. Moscow 2000, 65, 757–765.
- Culbertson, J.Y.; Kreider, R.B.; Greenwood, M.; Cooke, M.B. Effects of Beta-Alanine on Muscle Carnosine and Exercise Performance:A Review of the Current Literature. Nutrients 2010, 2, 75–98.
- Tanokura, M.; Tasumi, M.; Miyazawa, T. 1H Nuclear magnetic resonance studies of histidine-containing di- and tripeptides. Estimation of the effects of charged groups on the pKa value of the imidazole ring. Biopolymers 1976, 15, 393–401.
- Davey, C. The significance of carnosine and anserine in striated skeletal muscle. Arch. Biochem. Biophys. 1960, 89, 303–308.
- Mannion, A.F.; Jakeman, P.M.; Dunnett, M.; Harris, R.C.; Willan, P.L.T. Carnosine and anserine concentrations in the quadriceps femoris muscle of healthy humans. Eur. J. Appl Physiol. Occup. Physiol. 1992, 64, 47–50.
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and Pathophysiology of Carnosine. Physiol. Rev. 2013, 93, 1803–1845.
- Hoetker, D.; Chung, W.; Zhang, D.; Zhao, J.; Schmidtke, V.K.; Riggs, D.W.; Derave, W.; Bhatnagar, A.; Bishop, D.J.; Baba, S.P. Exercise alters and beta-alanine combined with exercise augments histidyl dipeptide levels and scavenges lipid peroxidation products in human skeletal muscle. J. Appl. Physiol. 1985 2018, 1767–1778, 125.
- Drozak, J.; Veiga-Da-Cunha, M.; Vertommen, D.; Stroobant, V.; Van Schaftingen, E. Molecular Identification of Carnosine Synthase as ATP-grasp Domain-containing Protein 1 (ATPGD1). J. Biol. Chem. 2010, 285, 9346–9356.
- Harding, J.W.; O’Fallon, J.V. The subcellular distribution of carnosine, carnosine synthetase, and carnosinase in mouse olfactory tissues. Brain Res. 1979, 173, 99–108.
- Ng, R.H.; Marshall, F.D.; Henn, F.A.; Sellström, Å. Metabolism of carnosine and homocarnosine in subcellular fractions and neuronal and glial cell-enriched fractions of rabbit brain. J. Neurochem. 1977, 28, 449–452.
- Deutsch, A.; Eggleton, P. The titration constants of anserine, carnosine and some related compounds. Biochem. J. 1938, 32, 209–211.
- Aldini, G.; Orioli, M.; Rossoni, G.; Savi, F.; Braidotti, P.; Vistoli, G.; Yeum, K.-J.; Negrisoli, G.; Carini, M. The carbonyl scavenger carnosine ameliorates dyslipidaemia and renal function in Zucker obese rats. J. Cell. Mol. Med. 2010, 15, 1339–1354.
- Baba, S.P.; Hoetker, J.D.; Merchant, M.; Klein, J.B.; Cai, J.; Barski, O.A.; Conklin, D.J.; Bhatnagar, A. Role of Aldose Reductase in the Metabolism and Detoxification of Carnosine-Acrolein Conjugates. J. Biol. Chem. 2013, 288, 28163–28179.
- Ihara, H.; Kakihana, Y.; Yamakage, A.; Kai, K.; Shibata, T.; Nishida, M.; Yamada, K.-I.; Uchida, K.; Yamda, K.-I. 2-Oxo-histidine–containing dipeptides are functional oxidation products. J. Biol. Chem. 2018, 294, 1279–1289.
- Baran, E.J. Metal complexes of carnosine. Biochem. Mosc. 2000, 65, 789–797.
- Gualano, B.; Everaert, I.; Stegen, S.; Artioli, G.G.; Taes, Y.; Roschel, H.; Achten, E.; Otaduy, M.C.; Júnior, A.H.L.; Harris, R.; et al. Reduced muscle carnosine content in type 2, but not in type 1 diabetic patients. Amino Acids 2011, 43, 21–24.
- Mong, M.C.; Chao, C.Y.; Yin, M.C. Histidine and carnosine alleviated hepatic steatosis in mice consumed high saturated fat diet. Eur. J. Pharmacol. 2011, 653, 82–88.
- Sauerhöfer, S.; Yuan, G.; Braun, G.S.; Deinzer, M.; Neumaier, M.; Gretz, N.; Floege, J.; Kriz, W.; Van Der Woude, F.; Moeller, M.J. L-Carnosine, a Substrate of Carnosinase-1, Influences Glucose Metabolism. Diabetes 2007, 56, 2425–2432.
- De Courten, B.; Jakubova, M.; De Courten, M.P.; Kukurova, I.J.; Vallova, S.; Krumpolec, P.; Valkovic, L.; Kurdiova, T.; Garzon, D.; Barbaresi, S.; et al. Effects of carnosine supplementation on glucose metabolism: Pilot clinical trial. Obesity Silver Spring 2016, 24, 1027–1034.
- Regazzoni, L.; De Courten, B.; Garzon, D.; Altomare, A.; Marinello, C.; Jakubova, M.; Vallova, S.; Krumpolec, P.; Carini, M.; Ukropec, J.; et al. A carnosine intervention study in overweight human volunteers: Bioavailability and reactive carbonyl species sequestering effect. Sci. Rep. 2016, 6, 27224.
- Teufel, M.; Saudek, V.; Ledig, J.-P.; Bernhardt, A.; Boularand, S.; Carreau, A.; Cairns, N.J.; Carter, C.J.; Cowley, D.J.; Duverger, D.; et al. Sequence Identification and Characterization of Human Carnosinase and a Closely Related Non-specific Dipeptidase. J. Biol. Chem. 2002, 278, 6521–6531.
- Orioli, M.; Vistoli, G.; Regazzoni, L.; Pedretti, A.; Lapolla, A.; Rossoni, G.; Canevotti, R.; Gamberoni, L.; Previtali, M.; Carini, M.; et al. Design, synthesis, ADME properties, and pharmacological activities of beta-alanyl-D-histidine (D-carnosine) prodrugs with improved bioavailability. Chem. Med. Chem. 2011, 6, 1269–1282.
- Menini, S.; Iacobini, C.; Ricci, C.; Scipioni, A.; Fantauzzi, C.B.; Giaccari, A.; Salomone, E.; Canevotti, R.; Lapolla, A.; Orioli, M.; et al. D-carnosine octylester attenuates atherosclerosis and renal disease in ApoE null mice fed a Western diet through reduction of carbonyl stress and inflammation. Br. J. Pharmacol. 2012, 166, 1344–1356.
- Everaert, I.; De Naeyer, H.; Taes, Y.; Derave, W. Gene expression of carnosine-related enzymes and transporters in skeletal muscle. Eur. J. Appl. Physiol. 2013, 113, 1169–1179.
- Scheuermann-Freestone, M.; Madsen, P.L.; Manners, D.; Blamire, A.M.; Buckingham, R.E.; Styles, P.; Radda, G.K.; Neubauer, S.; Clarke, K. Abnormal Cardiac and Skeletal Muscle Energy Metabolism in Patients With Type 2 Diabetes. Circulation 2003, 107, 3040–3046.
- Ingram, K.H.; Hill, H.; Moellering, D.; Hill, B.G.; Lara-Castro, C.; Newcomer, B.; Brandon, L.J.; Ingalls, C.P.; Penumetcha, M.; Rupp, J.C.; et al. Skeletal muscle lipid peroxidation and insulin resistance in humans. J. Clin. Endocrinol. Metab. 2012, 97, E1182–E1186.
- Heikkinen, S.; Pietilä, M.; Halmekytö, M.; Suppola, S.; Pirinen, E.; Deeb, S.S.; Jänne, J.; Laakso, M. Hexokinase II-deficient mice. Prenatal death of homozygotes without disturbances in glucose tolerance in heterozygotes. J. Biol. Chem. 1999, 274, 22517–22523.
- Rahimi, Y.; Camporez, J.P.G.; Petersen, M.C.; Pesta, D.; Perry, R.J.; Jurczak, M.J.; Cline, G.W.; Shulman, G.I. Genetic activation of pyruvate dehydrogenase alters oxidative substrate selection to induce skeletal muscle insulin resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 16508–16513.
- Avogaro, A.; Toffolo, G.; Miola, M.; Valerio, A.; Tiengo, A.; Cobelli, C.; Del Prato, S. Intracellular lactate- and pyruvate-interconversion rates are increased in muscle tissue of non-insulin-dependent diabetic individuals. J. Clin. Investig. 1996, 98, 108–115.