Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Tobacco is a known risk factor for lung cancer, and continued tobacco use is associated with poorer outcomes across multiple lung cancer treatment modalities including surgery, chemotherapy and radiation therapy. Less is known about the association of tobacco use and outcomes with immune checkpoint inhibitors (ICIs), which are becoming an important part of the treatment landscape in lung cancer, both in metastatic and curative settings.
- tobacco use
- immune checkpoint inhibitors
- lung cancer
1. Introduction
Lung cancer is the leading cause of cancer-related mortality, contributing to almost 1.8 million deaths worldwide in 2020 [1]. Non-small cell lung cancer (NSCLC) accounts for 80% of cases, and two thirds are diagnosed at an advanced stage. Five-year survival rates for early and advanced-stage NSCLC are 57% and 6%, respectively [2]. Tobacco smoking is a major preventable risk factor for lung cancer, accounting for over 87% of lung cancer deaths [3].
The development of targeted therapies and immune checkpoint inhibitors (ICIs) have drastically changed the treatment landscape for NSCLC, improving drug tolerance and treatment outcomes. Inhibitors against programmed cell death protein 1 (PD-1) block the interaction between PD-1 and its ligand (PD-L1), restoring T-cell function and anti-tumour activity [4]. Anti-PD-1 agents are currently used as monotherapy, in combination with chemotherapy or other checkpoint inhibitors, depending on the patient’s PD-L1 tumour expression [5].
Durable long-term survival benefit with ICIs occurs in approximately 20% of patients with advanced NSCLC [6]. Accurate biomarkers to predict ICI response among lung cancer patients is currently lacking. Beyond PD-L1 tumour proportion score (TPS), other biomarkers such as tumour mutation burden (TMB), specific tumour mutations in TP53 and KRAS genes, inflammatory signatures including gamma interferon signalling and tumour-infiltrating lymphocytes (TIL) density have also been explored to predict benefit from anti-PD-1 agents [7,8]. Interestingly, several studies and clinical experience have shown that patients that are current or former smokers appear to have improved outcomes with anti-PD-1 therapy compared to never smokers [9,10]. This has been observed irrespective of PD-L1 expression [11,12], and may be related to the etiology and biological differences between lung cancer in smokers and non-smokers.
2. Tobacco Smoking and the Immune System
A single cigarette contains over 7000 hazardous chemicals and 60 carcinogens, including polycyclic hydrocarbon carcinogens such as benzo[a]pyrene (BAP) and nicotine-derived nitrosoaminoketone [13,14]. Polycyclic hydrocarbons are responsible for the formation of bulky DNA adducts and their removal by nucleotide excision repair may result in DNA damage, tumorigenesis, and enhanced mutation burden [15,16,17]. This creates a distinct mutational signature in smokers (smoking signature, SS) characteristic of C>A transversions [18]. Tobacco smoking is also associated with the upregulation of PD-L1, which impairs the inflammatory response, allowing tumor cells to evade the immune system [19,20,21] (Figure 1).
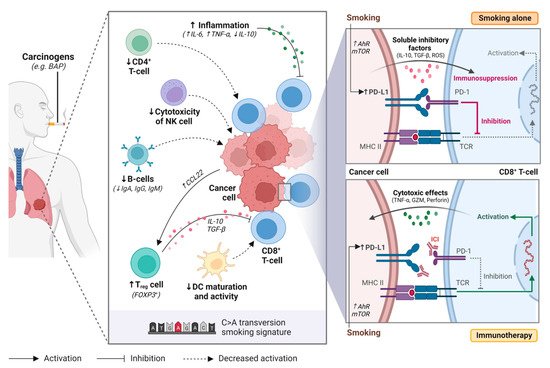
Figure 1. Tobacco smoking alters the immune and tumour microenvironment of lung cancer. Smoking may impair anti-tumour activity through over-activating the inflammatory immune response, upregulating regulatory T cell (Treg) function, decreasing natural killer (NK) cell cytotoxicity, downregulating B-cell activity and antibody production (i.e., IgA, IgG, IgM), and supressing dendritic cell (DC) maturation and activity. Smoking also increases programmed death-ligand 1 (PD-L1) expression on cancer cells through aryl hydrocarbon receptors (AhR)-related pathways or mTOR signaling, resulting in T-cell inactivation. Immune checkpoint inhibitors restore immune activation through disrupting the programmed death 1 (PD-1)/PD-L1 interaction. BAP, benzo[a]pyrene; NK cell, natural killer cell; Treg cell, regulatory T cell; DC, dendritic cell; Ahr, aryl hydrocarbon receptor; mTOR, mechanistic target of rapamycin; ICI, immune checkpoint inhibitor; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; MHC II, major histocompatibility complex II; TCR, T-cell receptor. Figure created with Biorender.com.
In healthy individuals, the PD-1/PD-L1 axis dampens the immune system to protect the host against autoimmunity. However, tumor cells use this same mechanism to evade the immune system [19]. Tobacco smoking is associated with high tumor PD-L1 expression and has a dose-dependent effect [19,21]. While the exact mechanism responsible for this phenomenon remains unclear, it has been hypothesized that tobacco carcinogens such as BAP may be responsible for upregulating PD-L1 expression [19]. BAP activation is mediated by aryl hydrocarbon receptors (AhR), that detoxify xenobiotics and regulate immune cell function [22]. Wang et al. observed that silencing AhR in mice decreased PD-L1 expression and prolonged survival [19]. Furthermore, treatment with anti-PD-1 agents suppressed tumor formation and significantly enhanced T-cell infiltration. In lung cancer patient samples, they observed higher AhR and PD-L1 expression in smokers compared to never smokers. Patients with high AhR-expressing tumors had greater clinical benefit with pembrolizumab, including in multivariable analysis [19].
Xiao et al. have proposed that mTOR signaling is responsible for stimulating PD-L1 expression through IL-6 [20]. When inhibiting mTOR using rapamycin, a significant reduction in PD-L1 expression was observed in experimental models. The addition of IL-6 recombinant protein to tobacco-cultured cell lines increased PD-L1 expression and partially restored tobacco-stimulated PD-L1 expression in rapamycin-treated cell lines. Together, these findings suggest that tobacco carcinogens play a role in increasing PD-L1 expression, and that these mechanisms may improve response to anti-PD-1 agents.
2.1. Smoking and Immune Cells
Chemicals found within tobacco cigarettes can impede the development, effector function and cytokine production of the immune system and alter the tumor immune microenvironment [23,24,25]. Through increasing the production of pro-inflammatory cytokines and reducing anti-inflammatory cytokine production, smoking induces an inflamed tumor microenvironment that promotes tumorigenesis and leads to chronic stimulation of T-cells with subsequent exhaustion and activation-induced cell death [23,26,27,28].
CD8+ tumor-infiltrating lymphocytes (TILs) are crucial for eliciting a direct cell-mediated antitumor immune response [29]. A combination of high CD8+ and low regulatory T-cell (Treg) infiltration is associated with prolonged survival in cancer patients [30,31]. High densities of tumor stromal infiltrating CD4+ and CD8+ T-cells have been significantly associated with smoking status in NSCLC [32] and other lung diseases [33]. However, other studies suggest no association between TIL density and smoking status [34,35]. Hiraoka et al. observed that simultaneously high densities of CD4+ and CD8+ T-cells have a synergistic effect and are associated with a favorable prognosis in lung cancer [36]. Despite the high abundance of CD8+ T-cells in the tumor stroma, they are functionally impaired and poorly responsive to T-cell activating stimuli [26,37]. This may be due to ineffective tumor-antigen presentation by dendritic cells (DC), reduced activity of CD4+ T-cells, and regulatory functions by Treg cells [26].
The highly inflammatory immune response associated with smoking also favors the accumulation and activation of DC [38,39]. However, smoking may also suppress DC function and maturation [26]. In tumor supernatants, Sharma et al. demonstrated that DCs had reduced surface expression of major histocompatibility complex (MHC) class I and II and co-stimulatory molecules (e.g., CD80 and CD86) in vitro, reducing their capacity to present tumor-specific antigens for T-cell activation [40]. Similar findings were observed in mice [27,41] and human DC cell lines [42], where exposure to cigarette smoking extracts hindered DC ability to stimulate and activate antigen-specific T-cells. Thus, cigarette smoking appears to have a profound impact on DC function and subsequent immunosuppression.
CD4+ T-cells are important for the initiation and maintenance of CD8+ T-cell activity [43]. Treg cells are a subset of CD4+ effector T-cells that maintain immunological homeostasis through immunosuppression [44]. Depletion of Treg cells has previously been demonstrated to augment anti-tumor T-cell activity [45,46]. Smoking status is associated with a low frequency of CD4+ T-cells [47,48] and high frequency of Treg cells [31]. Kinoshita et al. observed a high FOXP3/CD4 ratio in smokers with lung adenocarcinoma, where FOXP3 regulates Treg development, and suggested that this was an unfavorable prognostic factor [47]. Likewise, Sato et al. showed significant association between lung adenocarcinomas with a smoking signature (C > A transversions) and FOXP3+ T-cells [49]. These studies demonstrate that tumors with smoking signatures were associated low expression of CD4+ T-cells and high levels of Treg cells, leading to reduced tumor-infiltrating CD8+ T-cell activity and poor prognosis in NSCLC. Treg cells can also act on CD8+ T-cells indirectly through suppressing DC function [50], preventing proper antigen presentation to T-cells, leading to immunosuppression.
2.2. Smoking, Tumor Mutational Burden (TMB) and the Genomic Landscape
There is mounting evidence that smoking status and smoking signatures in tissues are associated with high somatic mutation rates, neoantigen production, and higher TMB [49,51,52,53,54,55]. There is conflicting evidence as to whether or not this is a dose-dependent effect. In NSCLC patients, Nagahashi et al. showed that current smokers without oncogenic driver mutations had the highest TMB in their samples, (p < 0.01), as compared to former or never smokers [53]. No differences in TMB were observed based on duration or intensity of tobacco exposure or pack years (PY). In contrast, Wang et al. observed that a doubling of smoking PY in NSCLC patients was associated with 1.11-fold increase in TMB (p < 0.001). Doubling of the number of months since quitting smoking was associated with a 0.95-fold decrease in TMB (p < 0.001) [54]. In summary, smoking is strongly associated with TMB, which may be associated with improved treatment outcomes with anti-PD- therapy [55,56].
The NSCLC mutational landscape in smoking and never smoking patients differs considerably. Tobacco smokers often have a tumor genomic profile characteristic of high frequencies of C > A transversions and defective DNA mismatch repair [18,54]. Specific signatures are associated with smoking-induced lung cancer. Nik-Zainal et al. demonstrated that exposure to BAP could induce similar genomic signatures [57]. Never smokers, on the other, have distinct genomic profiles, characteristic of spontaneous deamination of 5-methylcytosine and ultraviolet-induced mutations, respectively [54]. NSCLC patients with a smoking history or tumors with smoking-related signatures had better response rates and survival outcomes with anti-PD-1 treatments than never smokers or those with tumors without smoking-related signatures [51,58].
This entry is adapted from the peer-reviewed paper 10.3390/curroncol29090492
This entry is offline, you can click here to edit this entry!