Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, Research & Experimental
Pulmonary hypertension (PH) is a severe progressive lung disorder characterized by pulmonary vasoconstriction and vascular remodeling, culminating in right-sided heart failure and increased mortality.
- HIF
- hypoxia-inducible factor
- mitochondria
1. Introduction
Pulmonary hypertension (PH) is a chronic multifactorial pulmonary vascular disease (PVD), which presents in five different categories based on its characteristics [1,2]. According to the 6th World Symposium on Pulmonary Hypertension (WSPH) recommendations, two criteria are essential for the diagnosis of PH: mean pulmonary artery pressure (mPAP) > 20 mmHg and increased peripheral vascular resistance (PVR) ≥3 Wood Units (WU) [3]. A direct correlation exists between the product of PVR and cardiac output (CO), and the difference between mPAP and the pulmonary arterial wedge pressure (PAWP) so that PVR × CO = (mPAP − PAWP) [3]. Thus, the inclusion of PVR in the definition of pre-capillary PH is a crucial indicator of pulmonary vascular disease (PVD), as it accounts for the interplay between CO, mPAP, and PAWP.
Rare forms of PH include group I PH, pulmonary arterial hypertension (PAH), and group IV PH due to obstruction of the pulmonary artery, primarily as a result of chronic thromboembolic PH (CTEPH). The more common forms of PH include group II PH due to left-sided heart failure, group III PH caused by chronic lung disease and/or hypoxia, and group V PH precipitated by other miscellaneous factors [3,4]. Regardless of the etiology, the main characteristics of PH are vasoconstriction and severe remodeling of pulmonary vessels. The features of pulmonary vascular remodeling include thickening of the media, hyperproliferation of vascular cells, enhanced muscularity, increased migration, and increased inflammatory cell recruitment [5,6,7].
Exposure of the alveoli to regional hypoxia constricts the pulmonary vasculature to compensate for the diminished tissue perfusion and maintain adequate arterial oxygenation, a phenomenon known as hypoxic pulmonary vasoconstriction (HPV) [4,8,9,10]. Hypoxic stimulation of signaling mechanisms in the pre-capillary pulmonary arterial smooth muscle cells (PASMCs) is essential to HPV. Moreover, chronic exposure to hypoxia (in the scale of hours to days), hence prolonged activation of PASMCs, increases pulmonary vascular resistance (PVR) and contributes to pulmonary hypertension [4,11,12].
Although evidence implicates mitochondrial reactive oxygen species (ROS) in the development of hypoxia-induced PH, possibly via stabilization of the hypoxia-inducible factor-1 (HIF1), the direct role of ROS in the development of PH and thus the therapeutic potential of antioxidant treatment are controversial. Thus, this article addresses the role of mitochondrial regulation of HIF1α signaling in chronic hypoxia-induced PH.
2. Regulation of Hypoxia Inducible Factor (HIF) in Hypoxia
The adaptive mechanisms of vascular cells to chronic hypoxia are orchestrated mainly via the activity of the oxygen-dependent transcription factors known as hypoxia-inducible factors (HIFs) [13]. The work of Wang and colleagues [14] encouraged scientific efforts to uncover the regulatory role of HIFs on cellular homeostasis in response to hypoxia [5,15,16,17]. HIF-mediated signaling is the cornerstone of oxygen homeostasis that controls multiple hypoxia-responsive genes on the transcriptional level. HIF heterodimers comprise α and β subunits; both are transcription factors of the basic helix–loop–helix PAS family [14]. The β subunit (HIFβ) is constitutively expressed, independent of cellular oxygen availability, while the α subunits, e.g., HIF1α, HIF2α, and HIF3α, constitute the oxygen-sensitive element of HIF [14,18,19]. Moreover, normal tissue oxygen levels promote the hydroxylation and sbsequent degradatin of the α subunit, which is reversed under hypoxia [14,20].
The prolyl hydroxylase domain proteins (PHDs) are active under a normoxic settings. Hydroxylation of the proline amino acid residues (present in the oxygen-dependent degradation (ODD) domain) targets HIFα subunit for degradation by the proteasome system. PHDs use molecular oxygen (O2), Fe2+ cation, and 2-oxoglutarate to hydroxylate HIFα, which is then ubiquitinated via the ligase complex enzyme von Hippel–Lindau (VHL). Thus, the PHDs, along with HIF, work as an efficient cellular oxygen sensor that is switched off in hypoxic states and dictates HIFα stabilization and its dimerization with HIFβ, or its proteasomal degradation under normoxia [21,22] (Figure 1). Evidence shows that HIF1α remains stable upon deletion of the ODD domain even in normoxic cells, which favors its dimerization with the β-subunit and drives HIF-dependent signaling, independent of the O2 level [23,24].
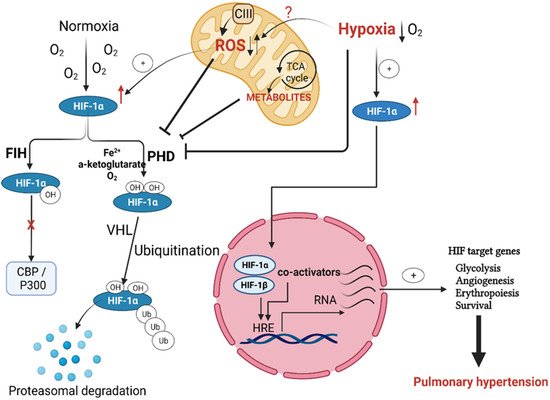
Figure 1. Regulation of hypoxia-inducible factor (HIF) activation in pulmonary hypertension. Both prolyl hydroxylases (PHD) and von Hipple–Lindau (VHL) hydroxylate HIF1α under normoxia, with subsequent targeting for proteasomal degradation. Factor-inhibiting HIF (FIH) controls HIF transactivation by impeding cofactor binding. Hypoxia, as well as mitochondria-produced ROS and metabolites, prevents HIF1 hydroxylation by PHD and subsequently allows its stabilization and nuclear translocation to form the active HIF transcription complex, which binds to its specific hypoxia response elements (HREs). The binding of this transcriptional complex to HREs activates HIF target genes essential to pulmonary hypertension initiation and progression.
Although the PHDs-mediated hydroxylation of the ODD domain is the primary determinant of HIF fate, deactivation of HIF-mediated gene transcription is also achieved by hydroxylation of the asparagine residue (Asp803) of the HIFα subunit C-terminal transactivation domain. The enzymatic activity of the factor inhibiting HIF (FIH) is responsible for this asparagine hydroxylation. Rather than the prolyl hydroxylation-facilitated proteasomal degradation, hydroxylation of Asp803 by FIH alters the conformation of HIF and prevents its binding with the transcriptional co-activators CBP and p300 [25,26] (Figure 1).
In hypoxia, reduced oxygen availability inhibits the HIFα subunit hydroxylation, leading to its accumulation, further translocation into the nucleus, and dimerization with the nuclear β-subunit forming a functional transcription unit with help of the transcriptional co-activators CBP and p300. This functional transcription complex binds to the hypoxia response elements (HREs) and allows the expression of hypoxia-specific genes [27,28]. Ultimately, the cellular response to results in cell survival, proliferation, and metabolic modulations such as repression of mitochondria respiration [29,30]. These HIF-mediated signals augment angiogenesis and erythropoiesis on the tissue- or organism-level [6,25,31] (Figure 1). Of the HIF target genes, erythropoietin (EPO) and the vascular endothelial growth factor (VEGF) are the primary mediators of the later effects. EPO is a glycoprotein hormone responsible for forming erythrocytes from their precursors via activating its EPO receptors, while VEGF is a primary stimulator of angiogenesis. These effects of EPO and VEGF improve oxygen supply to hypoxic tissue areas in the long term [30].
Besides hypoxia, previous studies showed the activation of HIF1 signaling by non-hypoxic conditions, such as the mechanical effects of vasoconstrictors, growth factors, cytokines, or hormones, which are upregulated in PH and induce transcription of HIF1α protein [32,33]. Moreover, the dysfunction of two key enzymes of the Krebs cycle: succinate and α-ketoglutarate dehydrogenases increases the accumulation of fumarate and succinate, which inhibit the activity of PHDs, and ultimately stabilize HIF1α [34,35].
Interestingly, HIF1α and HIF2α share a high level of structural similarity, whereas the HIF3α amino acid sequence is quite distinct. However, the three HIFα isoforms have similar O2-dependent regulation but differ in tissue expression and abundance [28]. HIF1α is expressed in most cell types, HIF2α can be found chiefly in the vascular endothelium and type II pneumocyte, and HIF3α is present in the cortex, heart, hippocampus, liver, lung, and kidney. Nevertheless, all HIFα subunits are subject to the exact degradation mechanisms [6,28].
3. HIF Signaling in Hypoxia-Induced PH
Extended exposure to low oxygen levels (chronic hypoxia) activates the transcription of HIF-dependent genes (Figure 2) that regulate the metabolism and proliferation of pulmonary vascular cells, blood vessel tone, and angiogenesis [18]. Hypoxia-induced changes in the PASMCs are critical to PH development after hypoxia exposure [36,37].
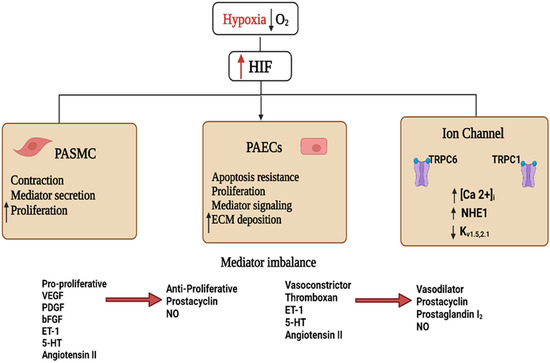
Figure 2. Schematic diagram showing the hypoxia-inducible factor 1 (HIF1)-activated mechanisms involved in pulmonary hypertension. Decreased blood O2 partial pressure (↓pO2 = hypoxia) acts as a signal in the pulmonary vasculature. Increased HIF stability and signaling during hypoxia activate cellular pathways culminating in vascular endothelial and smooth muscle dysfunction. These effects include increased secretion of vaso-modulators and extracellular matrix (ECM), depolarization of pulmonary artery vascular smooth muscle cells (PAVSMCs) via reduction of outward K+ and enhanced inward Ca2+ currents, and upregulated Na+/H+ exchanger isoform 1 (NHE1) function, increasing [Ca2+]i, contractility, and proliferation of PAVSMCs. The imbalance between vasodilator and vasoconstrictor signals and the altered cell proliferation orchestrates the pathological signaling events leading to vascular remodeling and the development of pulmonary hypertension. Abbreviations: bFGF, basic fibroblast growth factor; ET-1, endothelin 1; 5-HT, serotonin; NO, nitric oxide; PDGF, platelet-derived growth factor; PASMCS, pulmonary artery smooth muscle cells; PAECs, pulmonary artery endothelial cells; VEGF, vascular endothelial growth factor.
The role of HIF1α in altering pulmonary vascular contractility and remodeling has been studied extensively in mice with smooth muscle cells (SMCs)-specific HIF1α deletion. Specific deletion of HIF1α in SMCs in chronic hypoxic mice reduced pulmonary vascular remodeling and PH. However, hypertrophy of the right ventricle was not altered despite decreased pulmonary arterial pressures [38,39]. In contrast, others demonstrated that in mice exposed to normoxia and hypoxia, complete deletion of HIF1α in PASMCs increased the systolic pressure of the right ventricle and myosin light chain (MLC) phosphorylation. However, these mice showed a pulmonary artery muscularization phenotype comparable to controls. Thus, these results demonstrated the role of HIF1α in regulating vascular tone [40].
Under hypoxic conditions, PASMCs showed increased HIF1α-dependent expression of the micro-RNAs miR-9-1 and miR-9-3 [41] and inhibition of BMP signaling [42], which contributed to cell proliferation. Other HIF-regulated targets include genes that enhance oxidative stress, vascular tone, and mitochondrial fragmentation, as well as the activation of the renin-angiotensin-aldosterone system (RAAS) (Figure 2) [6,43].
Chronic hypoxia exposure of pulmonary artery endothelial cells (PAECs) resulted in different phenotypes with respect to angiogenesis, migration, and proliferation, which is largely defined by HIF isoforms [44,45,46]. For example, both HIF1/2-mediated signaling events in PAECs alter the expression glucose transporters 1/3 (GLUT1/3), hexokinase 1/2 (HK1/2), lactate dehydrogenase A (LDHA), and pyruvate dehydrogenase kinase 1 (PDK1) to regulate anaerobic glycolysis [47,48]. Furthermore, HIF2α in isolated PAECs activates the arginase-1- and arginase-2-dependent pathways, which reduce vascular NO homeostasis. Such effects contribute to the development of PH in chronic hypoxia-exposed mice. Moreover, loss of arginase-1 in PAECs inhibited the development of PH upon exposing the mice to chronic hypoxia [49].
Activation of HIF1α is involved in the contraction of the pulmonary vasculature via its regulation of ion channel expression. In hypoxia, HIF1α promotes the expression of the transient receptor potential channel members TRPC1 and TRPC6, leading to elevated PASMCs cytoplasmic Ca2+ concentrations, increasing the contractility and proliferation of isolated PASMCs (Figure 2) [50,51]. Additionally, hypoxia exposure reduces the expression of the voltage-gated potassium channels Kv1.5 and Kv2.1, causing a reduction in K+ current, subsequent membrane depolarization, voltage-gated calcium channel activation, and increased intracellular Ca2+. Further evidence on the impact of HIF1α activation in controlling both vascular tone and pulmonary vascular remodeling comes from studies showing that isolated HIF1α-deficient PASMCs display normal K+ currents following exposure to chronic hypoxia [52,53].
Akin to its effect on calcium homeostasis, the role of HIF1α in PH pathogenesis may also involve the dysregulation of pH homeostasis. Consistent with this hypothesis, both hypoxia and hypoxia-induced HIF1α upregulation activate the Na+/H+ exchanger isoform 1 (NHE1) (Figure 2) [53], leading to alkalinization of the intracellular pH favoring PASMCs proliferation [28,53]. On the other hand, inhibition of NHE attenuated the development of chronic hypoxia-induced PH and vascular remodeling [54].
Finally, chronic hypoxia exposure causes inflammation as an early consequence and an essential component in developing PH. The inflammatory targets of HIF activation include IL-6, nuclear factor kappaB (NF-κB), stromal cell-derived factor-1, and VEGF in PH patients and animal models [43,53,55,56]. Moreover, signaling pathways activated by inflammatory cytokines, such as NF-kB, can further trigger HIF1α activity and potentiate the signaling pathway mTOR/PI3K/Akt, a common downstream inflammation pathway, resulting in HIF stabilization and NF-κB activation [28,31,57]. With HIF1α, these inflammatory signaling events comprise central modulators of the energy metabolism towards aerobic glycolysis, a phenomenon which takes place in both cancer and PAH [58,59,60]. Thus, the ability of inflammatory signals to alter the signaling pathways in the pulmonary vasculature, possibly via initiation of mTOR, NF-κB, HIF signaling, and hypoxia-induced metabolic switch, merits further research.
4. Targeting HIF as a Potential Therapeutic Strategy in Pulmonary Hypertension
Many investigated compounds have now been utilized in the preclinical stage to assess their ability to target the HIF pathway therapeutically and determine hypoxic responses in the lung [22]. Most of these compounds showed promising results in reversing or inhibiting the incidence of PH in experimental models.
Multiple studies have pharmacologically targeted PHD2, HIF1α, or HIF2α as components of the HIF pathway in models of PH. These inhibitors, given by various routes of administration, attenuated or reversed PH in different rodent models of PH (hypoxia, MCT, and SuHx) [27]. For example, camptothecin and topotecan deactivate HIF1 at the level of mRNA expression, while celastramycin, 2-methoxyestradiol, and digoxin decrease protein synthesis [103].
In contrast, the YC-1 molecule targets protein accumulation and transcriptional regulation of the HIF axis. Moreover, apigenin and mAb AA98 have altered specific signaling molecules in HIF1 pathway regulation. Additionally, the link between HIF1α, NF-κB, and phosphatidylinositol 3-kinase (PI3K) signaling in PASMCs [114] and other vascular networks continues to gain interest [43]. A previous study demonstrated that caffeic acid phenethyl ester (CAPE), known to inhibit NF-κB, significantly suppressed the AKT/ERK/HIF1α signaling axis in MCT- or chronic hypoxia-induced animal models of PH. This study showed that the CAPE inhibited vascular remodeling by inhibiting HIF1α-induced activation of AKT and ERK signaling. Suppression of those signaling pathways by CAPE inhibited vascular cell proliferation and enhanced the apoptosis of PASMCs [113].
Other potential therapeutics include C76, which suppresses HIF2α at the level of mRNA, heterodimerization, and DNA binding [26]. Notably, C76 showed significant anti-remodeling effects in different animal models of PH [27,103], which manifested as attenuation of vascular muscularization, right-sided hypertrophy, and PAPs in hypoxia-exposed animals, suggesting that HIF2α inhibition could provide a promising therapeutic approach to attenuate hypoxia-induced PH [3,15,39]. Thus, therapeutic targeting of this route is receiving increased interest. Iron supplementation is another way to target the HIF system, as PHD activity is iron-dependent. Indeed, iron infusions reduce the rise in pulmonary arterial pressures in response to acute and chronic hypoxia [115,116].
Furthermore, considering the vital role of ROS in the PH pathogenesis, inhibition of mtROS release by the mitochondria-targeted antioxidant MitoQ [71] or via genetic approaches, e.g., AOX overexpression [98] or Cox4i2 disruption [11], showed inhibition of acute hypoxic pulmonary vasoconstriction but not chronic hypoxia-induced PH. Moreover, incubation with an antioxidant inhibited PH development when administered prior to the hypoxic exposure but not simultaneously [117]. Additionally, in previous animal studies, several antioxidant-rich substances have been utilized to slow the course of PH pathogenesis. Examples include allopurinol, SOD, pyrrolidine dithiocarbamate (PDTC), and sulfur dioxide [118,119]. However, using available non-selective antioxidants may impact the tricky balance that promotes homeostatic redox signaling or causes detrimental oxidative stress. As a result, mitochondria-targeted compounds that inhibit specific molecules implicated in ROS generation, such as RISP in complex III, may present successful PH treatments [80].
5. Conclusions
Isoforms of HIF play a critical role in the pathogenesis of hypoxia-induced PH through cellular and molecular signaling modulation that alters vasoconstriction, PASMC proliferation, and angiogenesis. HIF1 and HIF2 exert various and complementary effects upon exposure to hypoxia in both pulmonary vascular smooth muscle and endothelial cells. In addition, mitochondria-derived mechanisms promote HIF stabilization with subsequent progression and development of the PH disease. Mitochondria regulate acute and chronic responses to hypoxia exposure. Longer-term adaptation to hypoxia exposure is mediated by HIF activation via ROS and/or metabolite-induced PHD deactivation. However, the detailed upstream and downstream mechanisms of HIF signaling require further investigation. More research is needed to find new, effective, and selective inhibitors that target specific locations in the HIF pathway and subsequent new therapeutic approaches for PH treatment.
This entry is adapted from the peer-reviewed paper 10.3390/jcm11175219
This entry is offline, you can click here to edit this entry!