Cancer cachexia (CC) is a multifactorial syndrome characterized by a significant reduction in body weight that is predominantly caused by the loss of skeletal muscle and adipose tissue. Although the ill effects of cachexia are well known, the condition has been largely overlooked, in part due to its complex etiology, heterogeneity in mediators, and the involvement of diverse signaling pathways. For a long time, inflammatory factors have been the focus when developing therapeutics for the treatment of CC. Despite promising pre-clinical results, they have not yet advanced to the clinic. Developing new therapies requires a comprehensive understanding of how deregulated signaling leads to catabolic gene expression that underlies muscle wasting.
1. Introduction
The word cachexia comes from the Greek word
Kakos hexis meaning ‘bad condition’. Cachexia is a complex syndrome which manifests at late stages of several chronic diseases including Cancer, Chronic Kidney Disease (CKD), Chronic Obstructive Pulmonary Disease (COPD), AIDS, etc. [
1]. Cancer Cachexia (CC) is often recognized or assessed only when cancer patients show signs of a significant loss in body weight. It affects up to 80% of cancer patients at late stages and occurs earlier in patients with gastrointestinal and lung cancer. Indeed, 20–30% of cachectic tumor patients die due to CC rather than from the tumor itself [
2]. Despite being recognized as a devastating cancer-associated condition, cachexia remains an unmet medical need. This is partly due to the complexity of the syndrome and partly to the lack of proper guidelines to define and diagnose the condition. In 2011, Fearon et al. proposed a framework to define and classify CC as a multifactorial syndrome with an ongoing loss of skeletal muscle mass with or without loss of fat mass, which cannot be reversed by nutritional support. A loss in body weight of >5% over 6 months is classified as cachexia [
3]. While sarcopenia refers to the gradual age-related loss in muscle mass/strength, cachexia is associated with skeletal muscle loss due to illness/disease. In addition to dealing with the tumor itself, cancer patients suffer multiple problems including weight loss, anorexia, increased resting energy expenditure, metabolic changes and systemic inflammation [
4]. All these symptoms contribute to a reduced tolerance to cancer treatment, poor quality of life and increased mortality in patients.
2. Mediators of Skeletal Muscle Wasting
In general, most cancer patients suffer from anorexia (loss of appetite). Even sufficient nutritional support fails to reverse the progressive weight loss. The metabolic changes differ between anorexic and cachectic patients [
1,
5]. This indicates that reduced food intake alone is less likely to contribute to the complex muscle-wasting process.
Parabiosis studies in animal models provide evidence for the humoral mediation of CC [
6,
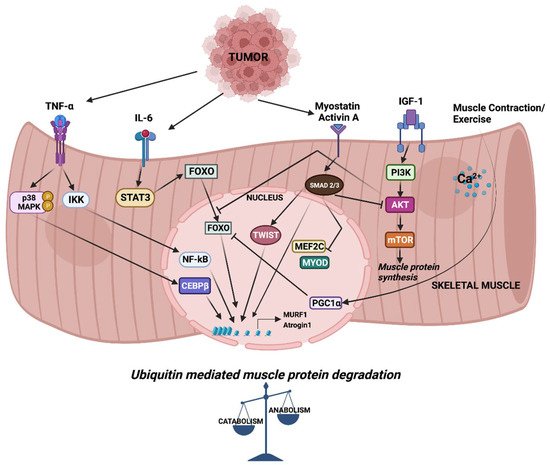
7]. These studies indicate that CC might be driven, at least in part, by circulating factors released by either host or tumor cells. Among these, inflammatory cytokines such as tumor necrosis factor alpha (TNFα), Interleukin-6 (IL-6) and transforming growth factor β (TGFβ) family members have been studied in detail. In CC, altered cytokine levels tilt the balance between muscle anabolic and catabolic genes towards catabolism, resulting in the degradation of muscle proteins. Cytokine-mediated signaling leading to the transcriptional regulation of muscle catabolic genes is critical in cachexia. In particular, the expression of the E3 ubiquitin ligase muscle RING finger containing protein 1 (MURF1) and muscle atrophy F box protein (MAFbx, also known as Atrogin-1) are central to ubiquitin-mediated myofibrillar protein degradation in cachectic skeletal muscle [
8]. Several signaling pathways converge to activate the MURF1 and Atrogin-1 mediated degradation of skeletal muscle proteins that causes muscle wasting (
Figure 1). Atrogin-1 controls the activity of the translation machinery protein eIF3f by ubiquitination [
9]. Silencing Atrogin-1 leads to the upregulation of MyoD and the downregulation of myostatin, a negative regulator of muscle mass [
10].
Figure 1. Signaling and downstream transcriptional response in skeletal muscle wasting. Cytokines released from tumor cells trigger signaling pathways which activate a cascade of transcription factors, leading to the expression of MURF1 and Atrogin-1. Increased expression of MURF1 and Atrogin-1 causes imbalance in muscle anabolic and catabolic processes.
A large body of evidence consistently points to the involvement of the ubiquitin proteasome system (UPS) in the degradation of muscle proteins in CC. In humans, increased ubiquitin mRNA expression and increased proteolytic activity is evident in the muscle of gastric cancer cachexia patients [
11,
12]. A number of animal models of cachexia also showed increased activity of proteasomes [
13]. Indeed, the inhibition of proteasome using MG132 in tumor-bearing mice leads to reduced cachexia [
14].
3. Epigenetic Regulation of Muscle Catabolic Genes
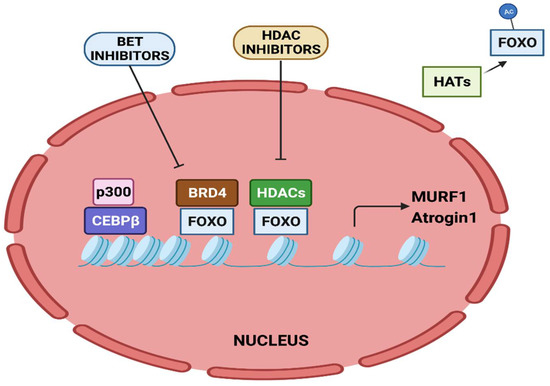
The activity of FOXO transcription factors is tightly regulated by epigenetic factors (
Figure 3). The acetylation of FOXO by p300-CBP acetyltransferase prevents nuclear localization and reduces its transcriptional activity [
112]. HDAC1 increases FOXO activity, causing the elevated expression of atrogenes including MURF1 and Atrogin-1, and leads to muscle atrophy [
113]. Trichostatin A (TSA), a HDAC inhibitor, counteracts unloading-induced muscle wasting. TSA treatment partly prevents the loss of type I and type IIa muscle fiber size in mice. Furthermore, HDAC4 interacts with and deacetylates FOXO3, leading to its activation, and thereby promotes denervation-induced muscle wasting in mice [
114]. N
6-methyladenosine (m
6A) is one of the most abundantly studied post-transcriptional modifications of eukaryotic mRNA. Using a denervation-induced muscle atrophy mouse model, the m
6A demethylase ALKB homologue 5 (ALKBH5) was shown to stabilize HDAC4 mRNA [
115].
Figure 3. Epigenetic regulation of MURF1 and Atrogin-1 transcription. Transcriptional activation of MURF1 and Atrogin-1 by FOXO and CEBPβ is regulated by epigenetic factors.
The bromodomain-containing BET protein promotes muscle wasting during cachexia. Administration of the BET inhibitor JQ1 in C26 tumor-bearing mice protects them from weight loss and muscle wasting. JQ1 administration orchestrates dual functions. It results in the loss of BRD4 at the promoters of catabolic genes. In addition, it results in reduced IL-6 levels, thereby restraining the IL-6/AMPK/FOXO3 activation of catabolic genes [
116].
Twist1, a key transcription factor involved in epithelial to mesenchymal transition, is known to play a role in cachexia. Twist1 plays a critical role in muscle protein degradation in CC and its expression is higher in the skeletal muscles of cachectic mice models. Its expression is induced by activin A via SMAD signaling that activates Atrogin-1 and MURF1, causing muscle proteolysis. The conditional deletion or pharmacological inhibition of Twist1 suppresses muscle protein degradation in CC [
117].
A loss in sarcomeric proteins, the contractile units of myofibrils, causes reduced muscle strength and induces wasting. SUMO-specific isopeptidase SENP3 determines sarcomere assembly by regulating the expression of sarcomeric contractile myosin heavy chain gene (MyHC-II). Under physiological conditions, SENP3 associates with the histone methyltransferase SETD7, causing its deSUMOylation. However, in cachectic muscle, SENP3 is degraded, which leads to the SUMOylation of SETD7. This allows the binding of SUV39H1 to MyHC-II, resulting in its repression, thereby causing disorganized sarcomeres [
118]. Interestingly, chemotherapeutic drugs such as etoposide and daunorubicin are responsible for chemotherapy-induced cachexia. These drugs destabilize SENP3, causing disrupted sarcomere organization through the dissociation of SETD7 and acetyltransferase p300, which leads to reduced acetylation and the downregulation of sarcomeric genes. These findings highlight the role of epigenetic factors and SENP3-regulated mechanisms in cachexia [
119].
Emerging evidence has demonstrated the association of non-coding RNAs with CC [
120]. Novel miRNAs which are involved in myogenesis and metabolism have been identified in cancer cachexia [
121]. Several miRNAs, including let-7d-3p, miR-345-5p, miR-532-5p, miR-378, miR-92a-3p and miR-21, are dysregulated in cachexia [
122]. Integrated miRNA and mRNA co-profiles during skeletal muscle wasting in cancer-induced cachexia showed that extracellular matrix (ECM) associated genes are post-transcriptionally regulated by miRNAs (such as miR-29a-3p) and atrophy-related transcription factors including NF-κB, STAT3 and FOXO [
123]. In addition, microarray data of long noncoding RNA (LncRNAs) in the adipose tissue showed that MALAT1 modulates adipose loss in CC by suppressing adipogenesis through PPAR-γ [
124].
ANAPC7 circular RNA (circRNA) is a novel tumor suppressor in pancreatic cancer. It functions through the CREB–miR-373–PHLPP2 axis and leads to AKT dephosphorylation and the downregulation of TGFβ and cyclinD1 to suppress muscle wasting and cachexia. PHLPP2 induces the dephosphorylation of CREB, thus, regulating cancer progression and cachexia [
125].
Tumor-secreted microvesicles contain an upregulated expression of miR-21, which induces myoblast apoptosis in CC through toll-like receptor 7/c-Jun N-terminal kinase-dependent pathway [
126]. Poly ADP ribose polymerase (PARP), a muscle metabolic enzyme, plays a role in cancer-induced cachexia. When compared to WT cachectic mice, Parp 1
-/- and Parp 2
-/- lung cancer cachectic mice show improvement in muscle fiber size, muscle strength and reduced tumor burden [
127].
Taken together, these studies highlight the interplay between epigenetic modifiers and transcription factors in regulating catabolic genes in muscle wasting. Given that CC does not involve somatic mutations, understanding the epigenetic regulation of catabolic genes might prove critical in the context of developing potential drug targets to treat muscle-wasting conditions.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14174258