Alzheimer’s disease (AD) is the most common neurodegenerative disorder and its prevalence is increasing. Very few drugs effectively reduce AD symptoms and thus, a better understanding of its pathophysiology is vital to design new effective schemes. Presymptomatic neuronal damage caused by the accumulation of Amyloid β peptide and Tau protein abnormalities remains a challenge, despite recent efforts in drug development. Importantly, therapeutic targets, biomarkers, and diagnostic techniques have emerged to detect and treat AD. Of note, the compromised blood-brain barrier (BBB) and peripheral inflammation in AD are becoming more evident, being harmful factors that contribute to the development of the disease. Perspectives from different pre-clinical and clinical studies link peripheral inflammation with the onset and progression of AD.
- Alzheimer’s disease
- blood-brain barrier
- blood-borne factors
- neurovascular unit
- exercise
1. Introduction
2. The Role of Aβ in AD
3. Tau and AD
4. The Synergy between Aβ and Tau
5. Neuroinflammation in Alzheimer’s Disease
6. The BBB Barrier in AD
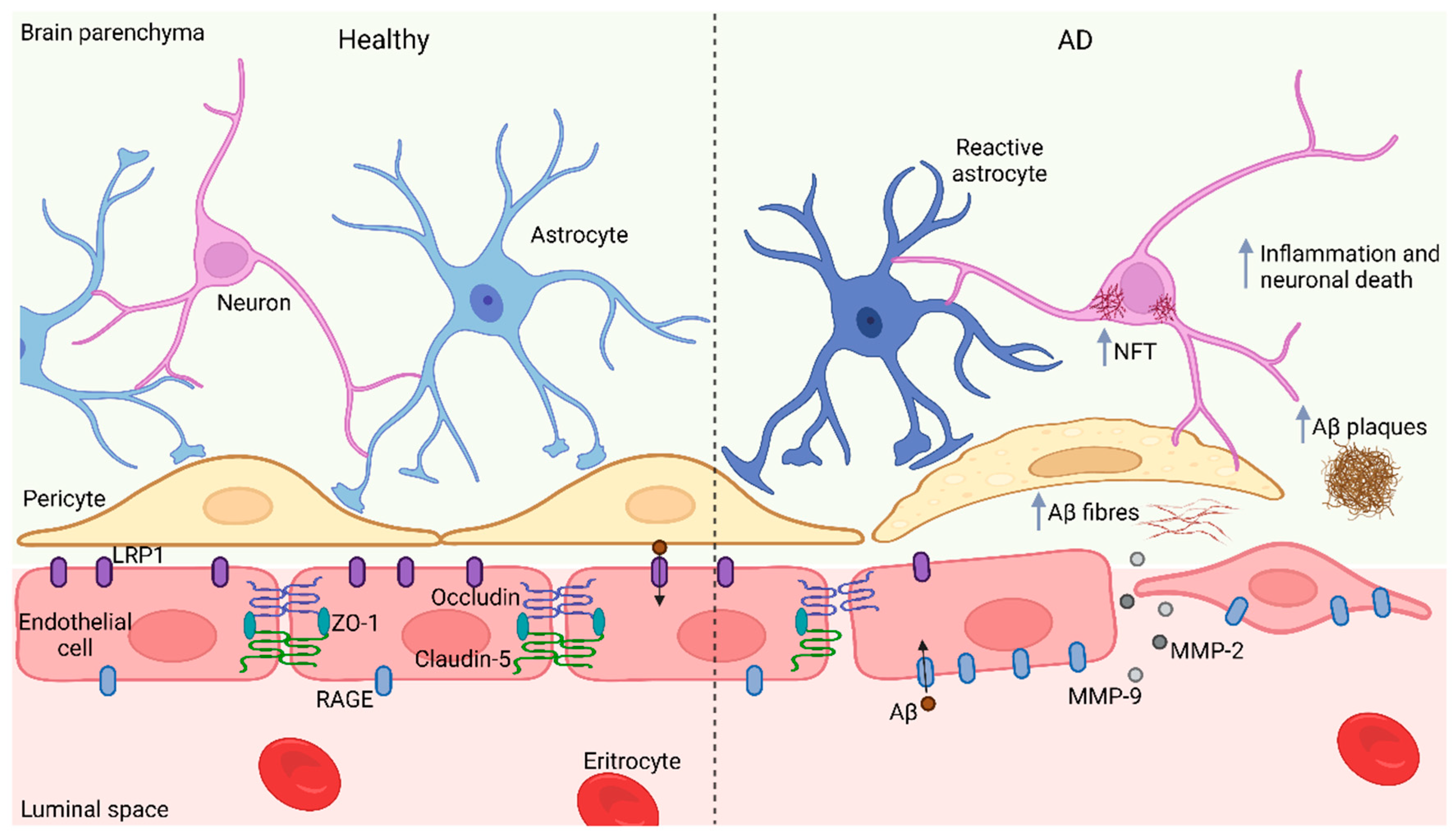
7. BBB Impairment: An Opportunity to Treat AD?
This entry is adapted from the peer-reviewed paper 10.3390/ijms231710136
References
- Dementia Statistics|Alzheimer’s Disease International (ADI). Available online: https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/ (accessed on 17 May 2022).
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213.
- Dhana, K.; Franco, O.H.; Ritz, E.M.; Ford, C.N.; Desai, P.; Krueger, K.R.; Holland, T.M.; Dhana, A.; Liu, X.; Aggarwal, N.T.; et al. Healthy Lifestyle and Life Expectancy with and without Alzheimer’s Dementia: Population Based Cohort Study. BMJ 2022, 377, e068390.
- Murphy, M.P.; Levine, H. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323.
- Ksiezak-Reding, H.; Liu, W.K.; Yen, S.H. Phosphate Analysis and Dephosphorylation of Modified Tau Associated with Paired Helical Filaments. Brain Res. 1992, 597, 209–219.
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The Precursor of Alzheimer’s Disease Amyloid A4 Protein Resembles a Cell-Surface Receptor. Nature 1987, 325, 733–736.
- Müller, U.C.; Deller, T.; Korte, M. Not Just Amyloid: Physiological Functions of the Amyloid Precursor Protein Family. Nat. Rev. Neurosci. 2017, 18, 281–298.
- Rice, H.C.; de Malmazet, D.; Schreurs, A.; Frere, S.; van Molle, I.; Volkov, A.N.; Creemers, E.; Vertkin, I.; Nys, J.; Ranaivoson, F.M.; et al. Secreted Amyloid-β Precursor Protein Functions as a GABA B R1a Ligand to Modulate Synaptic Transmission. Science 2019, 363, 123.
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An Overview of APP Processing Enzymes and Products. Neuromolecular Med. 2010, 12, 1.
- Ball, K.A.; Phillips, A.H.; Nerenberg, P.S.; Fawzi, N.L.; Wemmer, D.E.; Head-Gordon, T. Homogeneous and Heterogeneous Tertiary Structure Ensembles of Amyloid-β Peptides. Biochemistry 2011, 50, 7612–7628.
- Benilova, I.; Karran, E.; de Strooper, B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357.
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118.
- Ross, J. Science; Organization Science: Washington, DC, USA, 2022; Volume 358.
- Cable, J.; Holtzman, D.M.; Hyman, B.T.; Tansey, M.G.; Colonna, M.; Kellis, M.; Brinton, R.D.; Albert, M.; Wellington, C.L.; Sisodia, S.S.; et al. Alternatives to Amyloid for Alzheimer’s Disease Therapies—A Symposium Report. In Proceedings of the Annals of the New York Academy of Sciences; Blackwell Publishing Inc.: Hoboken, NJ, USA, 2020; Volume 1475, pp. 3–14.
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. 2019, 76, 915.
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The Physiological Roles of Tau and Aβ: Implications for Alzheimer’s Disease Pathology and Therapeutics. Acta Neuropathol. 2020, 140, 417.
- Shimada, H.; Kitamura, S.; Shinotoh, H.; Endo, H.; Niwa, F.; Hirano, S.; Kimura, Y.; Zhang, M.R.; Kuwabara, S.; Suhara, T.; et al. Association between Aβ and Tau Accumulations and Their Influence on Clinical Features in Aging and Alzheimer’s Disease Spectrum Brains: A PBB3-PET Study. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2017, 6, 11.
- Pittman, A.M.; Fung, H.C.; de Silva, R. Untangling the Tau Gene Association with Neurodegenerative Disorders. Hum. Mol. Genet. 2006, 15, R188–R195.
- Yamada, K.; Cirrito, J.R.; Stewart, F.R.; Jiang, H.; Finn, M.B.; Holmes, B.B.; Binder, L.I.; Mandelkow, E.M.; Diamond, M.I.; Lee, V.M.Y.; et al. In Vivo Microdialysis Reveals Age-Dependent Decrease of Brain Interstitial Fluid Tau Levels in P301S Human Tau Transgenic Mice. J. Neurosci. 2011, 31, 13110–13117.
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of Tau Protein in Alzheimer’s Disease: The Prime Pathological Player. Int. J. Biol. Macromol. 2020, 163, 1599–1617.
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210.
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Guerrero-Muñoz, M.J.; Jackson, G.R.; Kayed, R. Preparation and Characterization of Neurotoxic Tau Oligomers. Biochemistry 2010, 49, 10039–10041.
- Mattsson-Carlgren, N.; Andersson, E.; Janelidze, S.; Ossenkoppele, R.; Insel, P.; Strandberg, O.; Zetterberg, H.; Rosen, H.J.; Rabinovici, G.; Chai, X.; et al. Aβ Deposition Is Associated with Increases in Soluble and Phosphorylated Tau That Precede a Positive Tau PET in Alzheimer’s Disease. Sci. Adv. 2020, 6, eaaz2387.
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Review Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192.
- Small, S.A.; Duff, K. Linking Aβ and Tau in Late-Onset Alzheimer’s Disease: A Dual Pathway Hypothesis. Neuron 2008, 60, 534–542.
- Zheng, W.-H.; Bastianetto, S.; Mennicken, F.; Ma, W.; Kar, S. Amyloid L Peptide Induces Tau Phosphorylation and Loss Of Cholinergic Neurons In Rat Primary Septal Cultures. Neuroscience 2002, 115, 201–211.
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.-C.; Quinlan, M.; Wisniewski, H.M.; Bindert, L.I. Abnormal Phosphorylation of the Microtubule-Associated Protein X (Tau) in Alzheimer Cytoskeletal Pathology (Alzheimer Disease/Neurofibrillary Tangles/Paired-Helical Filaments/Microtubules). Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917.
- Silva, D.F.F.; Esteves, A.R.; Oliveira, C.R.; Cardoso, S.M. Mitochondria: The Common Upstream Driver of Amyloid-and Tau Pathology in Alzheimer´s Disease. Curr. Alzheimer Res. 2011, 8, 563.
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Dé Ric Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525.
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired Mitochondrial Dynamics and Abnormal Interaction of Amyloid Beta with Mitochondrial Protein Drp1 in Neurons from Patients with Alzheimer’s Disease: Implications for Neuronal Damage. Hum. Mol. Genet. 2011, 20, 2495–2509.
- Iijima, K.; Gatt, A.; Iijima-Ando, K. Tau Ser262 Phosphorylation Is Critical for Aβ42-Induced Tau Toxicity in a Transgenic Drosophila Model of Alzheimer’s Disease. Hum. Mol. Genet. 2010, 19, 2947–2957.
- Akama, K.T.; van Eldik, L.J. Beta-Amyloid Stimulation of Inducible Nitric-Oxide Synthase in Astrocytes Is Interleukin-1beta- and Tumor Necrosis Factor-Alpha (TNFalpha)-Dependent, and Involves a TNFalpha Receptor-Associated Factor- and NFkappaB-Inducing Kinase-Dependent Signaling Mechanism. J. Biol. Chem. 2000, 275, 7918–7924.
- Griffin, W.S.T.; Sheng, J.G.; Roberts, G.W.; Mrak, R.E. Interleukin-1 Expression in Different Plaque Types in Alzheimer’s Disease: Significance in Plaque Evolution. J. Neuropathol. Exp. Neurol. 1995, 54, 276–281.
- Mrak, R.E.; Griffin, W.S.T. Common Inflammatory Mechanisms in Lewy Body Disease and Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2007, 66, 683–686.
- Mrak, R.E.; Sheng, J.G.; Griffin, W.S.T. Glial Cytokines in Alzheimer’s Disease: Review and Pathogenic Implications. Hum. Pathol 1995, 26, 816–823.
- Tuppo, E.E.; Arias, H.R. The Role of Inflammation in Alzheimer’s Disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305.
- Nazem, A.; Sankowski, R.; Bacher, M.; Al-Abed, Y. Rodent Models of Neuroinflammation for Alzheimer’s Disease. J. Neuroinflamm. 2015, 12, 1–15.
- Saito, T.; Saido, T.C. Neuroinflammation in Mouse Models of Alzheimer’s Disease. Clin. Exp. Neuroimmunol. 2018, 9, 211.
- Gomez-Nicola, D.; Boche, D. Post-Mortem Analysis of Neuroinflammatory Changes in Human Alzheimer’s Disease. Alzheimers Res. Ther. 2015, 7, 1–8.
- Knezevic, D.; Mizrahi, R. Molecular Imaging of Neuroinflammation in Alzheimer’s Disease and Mild Cognitive Impairment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 123–131.
- Zimmer, E.R.; Leuzy, A.; Benedet, A.L.; Breitner, J.; Gauthier, S.; Rosa-Neto, P. Tracking Neuroinflammation in Alzheimer’s Disease: The Role of Positron Emission Tomography Imaging. J. Neuroinflamm. 2014, 11, 120.
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive Innate Immune Gene Activation Accompanies Brain Aging, Increasing Vulnerability to Cognitive Decline and Neurodegeneration: A Microarray Study. J. Neuroinflamm. 2012, 9, 179.
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a Central Mechanism in Alzheimer’s Disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590.
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139, 136–153.
- Grammas, P.; Martinez, J.; Sanchez, A.; Yin, X.; Riley, J.; Gay, D.; Desobry, K.; Tripathy, D.; Luo, J.; Evola, M.; et al. A New Paradigm for the Treatment of Alzheimer’s Disease: Targeting Vascular Activation. J. Alzheimer’s Dis. 2014, 40, 619–630.
- Grammas, P. Neurovascular Dysfunction, Inflammation and Endothelial Activation: Implications for the Pathogenesis of Alzheimer’s Disease. J. Neuroinflamm. 2011, 8, 26.
- Ros-Bernal, F.; Hunot, S.; Herrero, M.T.; Parnadeau, S.; Corvol, J.C.; Lu, L.; Alvarez-Fischer, D.; Carrillo-de Sauvage, M.A.; Saurini, F.; Coussieu, C.; et al. Microglial Glucocorticoid Receptors Play a Pivotal Role in Regulating Dopaminergic Neurodegeneration in Parkinsonism. Proc. Natl. Acad. Sci. USA 2011, 108, 6632–6637.
- Herrero, M.T.; Estrada, C.; Maatouk, L.; Vyas, S. Inflammation in Parkinson’s Disease: Role of Glucocorticoids. Front. Neuroanat. 2015, 9, 32.
- Faden, A.I.; Loane, D.J. Chronic Neurodegeneration after Traumatic Brain Injury: Alzheimer Disease, Chronic Traumatic Encephalopathy, or Persistent Neuroinflammation? Neurotherapeutics 2015, 12, 143–150.
- McCombe, P.A.; Henderson, R.D. The Role of Immune and Inflammatory Mechanisms in ALS. Curr. Mol. Med. 2011, 11, 246–254.
- Haase, S.; Linker, R.A. Inflammation in Multiple Sclerosis. Ther. Adv. Neurol. Disord 2021, 14.
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a Missense Mutation in the Amyloid Precursor Protein. Lett. Nat. 1991, 349, 704–706.
- Efthymiou, A.G.; Goate, A.M. Late Onset Alzheimer’s Disease Genetics Implicates Microglial Pathways in Disease Risk. Mol. Neurodegener. 2017, 12, 1–12.
- Xie, C.; Aman, Y.; Frank, J.; Donate-Lagartos, M.J.; Gudmundsrud, R.; Čechová, K.; Shi, L.; Vyhnalek, M.; Fang, E.F. Autophagic Processes in Early- and Late-Onset Alzheimer’s Disease. Autophagy Health Dis. 2022, 287–299.
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s Disease. Nat. Rev. Dis. Prim. 2015, 1, 15056.
- Xie, J.; van Hoecke, L.; Vandenbroucke, R.E. The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front. Immunol. 2022, 12, 5731.
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185.
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood–Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 1–24.
- Rhea, E.M.; Banks, W.A. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front. Neurosci. 2019, 13, 521.
- Zlokovic, B.V. Neurovascular Pathways to Neurodegeneration in Alzheimer’s Disease and Other Disorders. Nat. Rev. Neurosci. 2011, 12, 723–738.
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The Blood-Brain Barrier: An Engineering Perspective. Front. Neuroeng. 2013, 6, 7.
- Chowdhury, E.A.; Noorani, B.; Alqahtani, F.; Bhalerao, A.; Raut, S.; Sivandzade, F.; Cucullo, L. Understanding the Brain Uptake and Permeability of Small Molecules through the BBB: A Technical Overview. J. Cereb. Blood Flow Metab. 2021, 41, 1797–1820.
- Bernardo-Castro, S.; Sousa, J.A.; Brás, A.; Cecília, C.; Rodrigues, B.; Almendra, L.; Machado, C.; Santo, G.; Silva, F.; Ferreira, L.; et al. Pathophysiology of Blood–Brain Barrier Permeability Throughout the Different Stages of Ischemic Stroke and Its Implication on Hemorrhagic Transformation and Recovery. Front. Neurol. 2020, 11, 594672.
- Carvey, P.M.; Hendey, B.; Monahan, A.J. The Blood Brain Barrier in Neurodegenerative Disease: A Rhetorical Perspective. J. Neurochem. 2009, 111, 291.
- Pardridge, W.M. Drug Transport across the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959.
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological Blood–Brain Transport Is Impaired with Age by a Shift in Transcytosis. Nature 2020, 583, 425–430.
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-Brain Barrier Breakdown in Alzheimer Disease and Other Neurodegenerative Disorders. Nat. Rev. Neurol. 2018, 14, 133–150.
- Wisniewski, H.M.; Kozlowski, P.B. Evidence for Blood-Brain Barrier Changes in Senile Dementia of the Alzheimer Type (SDAT). Ann. N. Y. Acad. Sci. 1982, 396, 119–129.
- Wisniewski, H.M.; Vorbrodt, A.W.; Wegiel, J. Amyloid Angiopathy and Blood-Brain Barrier Changes in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 1997, 826, 161–172.
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78.
- Van De Haar, H.J.; Burgmans, S.; Jansen, J.F.A.; Van Osch, M.J.P.; Van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535.
- Oikari, L.E.; Pandit, R.; Stewart, R.; Cuní-López, C.; Quek, H.; Sutharsan, R.; Rantanen, L.M.; Oksanen, M.; Lehtonen, S.; de Boer, C.M.; et al. Altered Brain Endothelial Cell Phenotype from a Familial Alzheimer Mutation and Its Potential Implications for Amyloid Clearance and Drug Delivery. Stem Cell Rep. 2020, 14, 924–939.
- Takeda, S.; Sato, N.; Ikimura, K.; Nishino, H.; Rakugi, H.; Morishita, R. Increased Blood-Brain Barrier Vulnerability to Systemic Inflammation in an Alzheimer Disease Mouse Model. Neurobiol. Aging 2013, 34, 2064–2070.
- Wang, D.; Chen, F.; Han, Z.; Yin, Z.; Ge, X.; Lei, P. Relationship Between Amyloid-β Deposition and Blood–Brain Barrier Dysfunction in Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 695479.
- Erickson, M.A.; Banks, W.A. Blood-Brain Barrier Dysfunction as a Cause and Consequence of Alzheimer’s Disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513.
- Wu, Y.C.; Sonninen, T.M.; Peltonen, S.; Koistinaho, J.; Lehtonen, Š. Blood–Brain Barrier and Neurodegenerative Diseases—Modeling with Ipsc-derived Brain Cells. Int. J. Mol. Sci. 2021, 22, 7710.
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The Vascular Basement Membrane in the Healthy and Pathological Brain. J. Cereb. Blood Flow Metab. 2017, 37, 3300–3317.
- Nguyen, B.; Bix, G.; Yao, Y. Basal Lamina Changes in Neurodegenerative Disorders. Mol. Neurodegener. 2021, 16, 1–25.
- Hohsfield, L.A.; Humpel, C. Migration of Blood Cells to β-Amyloid Plaques in Alzheimer’s Disease. Exp. Gerontol. 2015, 65, 8–15.
- Pietronigro, E.; Zenaro, E.; Constantin, G. Imaging of Leukocyte Trafficking in Alzheimer’s Disease. Front. Immunol. 2016, 7, 1.
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone Marrow-Derived Microglia Play a Critical Role in Restricting Senile Plaque Formation in Alzheimer’s Disease. Neuron 2006, 49, 489–502.
- Zenaro, E.; Pietronigro, E.; Bianca, V.d.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils Promote Alzheimer’s Disease-like Pathology and Cognitive Decline via LFA-1 Integrin. Nat. Med. 2015, 21, 880–886.
- Michaud, J.P.; Bellavance, M.A.; Préfontaine, P.; Rivest, S. Real-Time in Vivo Imaging Reveals the Ability of Monocytes to Clear Vascular Amyloid Beta. Cell Rep. 2013, 5, 646–653.
- Kook, S.Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. Aβ1–42-RAGE Interaction Disrupts Tight Junctions of the Blood-Brain Barrier via Ca2+-Calcineurin Signaling. J. Neurosci. 2012, 32, 8845–8854.
- Middeldorp, J.; Lehallier, B.; Villeda, S.A.; Miedema, S.S.M.; Evans, E.; Czirr, E.; Zhang, H.; Luo, J.; Stan, T.; Mosher, K.I.; et al. Preclinical Assessment of Young Blood Plasma for Alzheimer Disease. JAMA Neurol. 2016, 73, 1325–1333.
- Sha, S.J.; Deutsch, G.K.; Tian, L.; Richardson, K.; Coburn, M.; Gaudioso, J.L.; Marcal, T.; Solomon, E.; Boumis, A.; Bet, A.; et al. Safety, Tolerability, and Feasibility of Young Plasma Infusion in the Plasma for Alzheimer Symptom Amelioration Study: A Randomized Clinical Trial. JAMA Neurol. 2019, 76, 35–40.
- De Miguel, Z.; Khoury, N.; Betley, M.J.; Lehallier, B.; Willoughby, D.; Olsson, N.; Yang, A.C.; Hahn, O.; Lu, N.; Vest, R.T.; et al. Exercise Plasma Boosts Memory and Dampens Brain Inflammation via Clusterin. Nature 2021, 600, 494–499.
- Horowitz, A.M.; Fan, X.; Bieri, G.; Smith, L.K.; Sanchez-Diaz, C.I.; Schroer, A.B.; Gontier, G.; Casaletto, K.B.; Kramer, J.H.; Williams, K.E.; et al. Blood Factors Transfer Beneficial Effects of Exercise on Neurogenesis and Cognition to the Aged Brain. Science 2020, 369, 167–173.
- Meng, Q.; Lin, M.S.; Tzeng, I.S. Relationship Between Exercise and Alzheimer’s Disease: A Narrative Literature Review. Front. Neurosci. 2020, 14, 131.
- Zhou, S.; Chen, S.; Liu, X.; Zhang, Y.; Zhao, M.; Li, W. Physical Activity Improves Cognition and Activities of Daily Living in Adults with Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Int. J. Environ. Res. Public Health 2022, 19, 1216.
- Caserta, M.T.; Caccioppo Gregory Lapin, D.D.; Ragin, A.; Groothuis, D.R. Blood-Brain Barrier Integrity in Alzheimer’s Disease Patients and Elderly Control. Subjects. J. Neuropsychiatry Clin. Neurosci. 1998, 10, 78–84.
- Schlageter, N.L.; Carson, R.E.; Rapoport, S.I. Examination of Blood—Brain Barrier Permeability in Dementia of the Alzheimer Type with EDTA and Positron Emission Tomography. J. Cereb. Blood Flow Metab. 1987, 7, 1–8.
- Starr, J.M.; Farrall, A.J.; Armitage, P.; McGurn, B.; Wardlaw, J. Blood-Brain Barrier Permeability in Alzheimer’s Disease: A Case-Control MRI Study. Psychiatry Res. Neuroimaging 2009, 171, 232–241.
- Pardridge, W.M. Treatment of Alzheimer’s Disease and Blood–Brain Barrier Drug Delivery. Pharmaceuticals 2020, 13, 1–25.