Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Ovarian cancer is one of the most lethal gynaecological malignancies worldwide. Despite high success rates following first time treatment, this heterogenous disease is prone to recurrence. Oncogenic activity of receptor tyrosine kinases is believed to drive the progression of ovarian cancer.
- ovarian cancer
- tyrosine kinases
- platinum resistance
1. Ovarian Cancer: Current Understanding and Treatment
Ovarian cancer (OC) is a leading cause of cancer-related death in women with the second highest mortality rate of gynaecological cancers worldwide [1]. In 2020, an estimated 313,959 cases were diagnosed worldwide, accounting for 3.4% of all new cases of cancer in women and an age-standardised incidence rate of 6.6 per 100,000 [1]. In the same year 207,252 deaths were recorded, accounting for 4.7% of women cancer related deaths with an age-standardised rate of 4.2 per 100,000 [1].
Diagnosis of OC follows the guidelines suggested by the International Federation of Gynaecology and Obstetrics (FIGO), consisting of a four-level staging system based on tumour location, histological profile, and level of metastatic activity [2]. Because early-stage diseases are often asymptomatic, around 75% of cases are only diagnosed at advanced stages III and IV, where tumours have disseminated and invaded the abdominal cavity beyond the pelvic region [2][3]. The highly invasive nature of OC results in the progression of the disease beyond the ovary, to the surrounding peritoneum, regional lymph nodes, and other organs within the peritoneal cavity. Even at an advanced stage of cancer, most common symptoms are nonspecific and include abdominal discomfort and bloating [3][4].
An estimated 85–90% of OC are of epithelial origin [5]. These are multicentric in nature and potentially include mesothelial invaginations, endosalpingiosis, and pelvic peritoneum. OC is highly heterogeneous and can be classed into subtypes based on histological profiles [2]. The most common form of OC is high grade serous ovarian carcinoma (HGSC), which is frequently studied in clinical trials. Other known histological subtypes of OC include clear cell, endometrioid, and mucinous carcinomas, as well as low-grade serous ovarian cancer (LGSC) [2][6]. Zhou et al., have suggested that histological subtypes are correlated with survival outcomes in patients. They found that 5-year overall survival was lower in patients with mucinous and clear cell subtypes (14.2% and 18.8%, respectively) as compared to HGSC and endometrioid (28.1% and 38.6%, respectively) [7].
Approximately half of the tumours also carry gene mutations causing defects in homologous recombination [8]. The mutational profiles of every histological subtype are different: HGSC usually carries TP53 mutations, LGSC and mucinous have increased in BRAF and RAS mutations, while endometrioid and ovarian clear cell carcinoma (OCCC) have mutations in ARID1A and PIK3CA [9][10]. Furthermore, transcriptomic analyses have demonstrated additional molecular subtypes of HGSC into mesenchymal, immunoreactive, differentiated, and proliferative [11][12], although the robustness and clinical utility of molecular subtyping remains obscure [11][13][14].
The standard first line treatment for OC involves the combination of surgical intervention and chemotherapy-based treatment [15]. Surgical intervention involves the practice of debulking, removing macroscopic tumoral deposits where possible to completely resect visible disease. Although the accuracy of tumour removal is often a strong predictor for overall survival (OS) [16], 50–60% of patients with advanced OC will require further treatment [17]. The common therapeutics administered are carboplatin and paclitaxel. Platinum/taxane chemotherapy is administered intravenously following debulking surgery over six cycles every 21 days [15]. If deemed fit, a patient may also be exposed to intraperitoneal chemotherapy [18].The late presentation and diagnosis, as well as the heterogeneous nature of OC results in the overall 5-year survival rate of 30–45% [19][20][21].
Despite the high success of first line treatment, 70% of advanced stage patients experience a relapse [22]. The classification of recurrence is based on the platinum-free interval (PFI), ranging from highly sensitive to platinum refractory, based on time taken for new tumours to occur post chemotherapy [15]. Platinum sensitive patients (>6-month period) can undergo further rounds of chemotherapy treatment. For platinum resistant and refractory patients, there is currently no evidence-based standard for a second line of treatment. These patients can be considered for experimental therapy in clinical trials settings [23]. Combination of chemotherapy and other cytotoxic agents has been clinically used to prolong this progression-free period [15]. Whilst the exact mechanism for the progression of OC during treatment has yet to be established, it is widely accepted that the cell signalling pathways of receptor tyrosine kinases may impact and influence the dynamic nature of these malignant cells [24]. With limited effective treatment available for chemoresistant or refractory patients, there is a critical need for the establishment of therapeutic agents. Small molecule inhibitors such as tyrosine kinase inhibitors (TKIs) and monoclonal antibodies (mAb) targeting tyrosine kinase have been implemented in the maintenance of tumour sensitivity and in cytoreduction in a variety of cancers including breast cancer [25], gastrointestinal stromal tumours, and renal cell carcinomas [26]. Several therapeutics of this nature have been trialled in preclinical and clinical settings to determine their efficacy and adverse effects in the treatment of OC in both single agent and combination usage.
2. Receptor Tyrosine Kinases
Receptor tyrosine kinases (RTKs) are enzymes tasked with the mediation of intercellular communication throughout the body [27]. RTKs exhibit their mode of action through the transfer of a phosphate group from ATP to one or more tyrosine residues, resulting in the conformational change of the target proteins [28][29]. Although tightly regulated under normal homeostatic conditions, RTK activity in cancer is highly dynamic, with altered functional roles due to mutations and overexpression [28]. The change in RTK function has been linked to malignancy in numerous cancers, including OC. The oncogenic activity of RTKs is assumed to be essential in the proliferation and progression of OC tumours [30][31]. RTKs rarely act in isolation, forming intricate co-activation networks with other TKs, diversifying signalling outcomes and preparing for ready adaptation should a pathway face interference [32]. This intercellular crosstalk is modified in cancers, with the plasticity of signalling demonstrated with chemoresistance and disease progression under therapeutic conditions [32][33]. Oncogenic activation of RTKs and their intermediates typically occurs through one of the following mechanisms: autocrine activation of RTK signalling (Figure 1a), chromosomal rearrangements (Figure 1b), duplication of tyrosine kinase domain (Figure 1c), gain of function mutations (Figure 1d), or genomic amplifications (Figure 1e) [24][27].
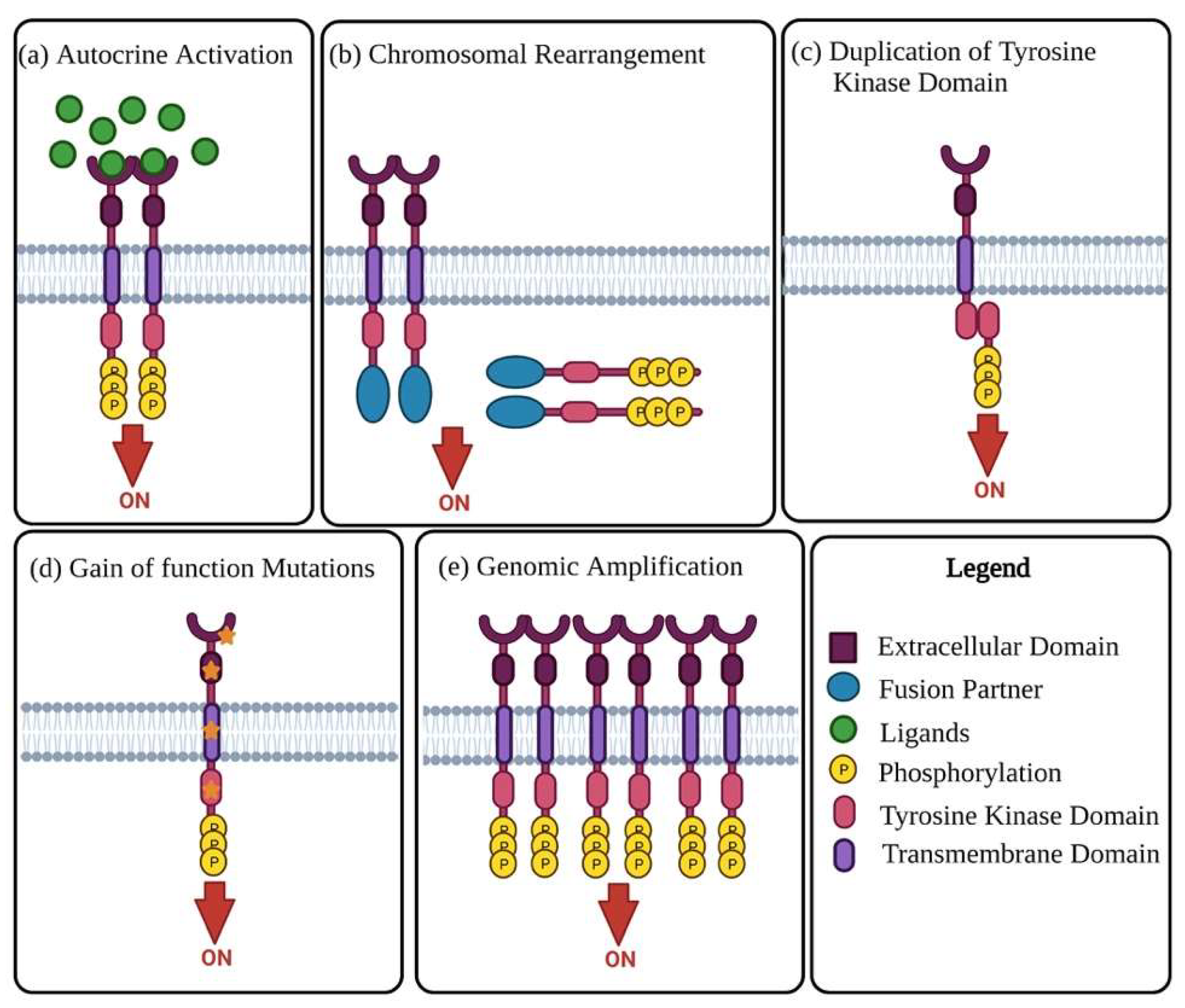
Figure 1. Mechanisms of oncogenic RTK activation. (a) Visual representation of autocrine activated RTK signalling. Increased ligand production by cancer cells or the tumour microenvironment causes activation of RTK signalling, leading to increased kinase activity and phosphorylation of the C-terminal tail of the receptor. (b) Chromosomal rearrangement results in the creation of a hybrid fusion oncoprotein composing partly of the TK and fusion partner. These RTK fusion partners are often cytoplasmic or membrane bound proteins depending on the position of the genomic cut-off point. The rearrangement results in deletion of regulatory domains, which then causes tyrosine kinase activation. (c) Duplication of tyrosine kinase domain could result in the formation of an intramolecular dimer and activation of RTK, in the absence of ligands. (d) Diagrammatic illustration of probable gain-of-function mutations in several RTK subdomains. Mutations in these regions lead to constitutive activation of the RTK, more often than not in the absence of ligands. (e) Visual representation of RTK genomic amplification is frequently a consequence of the genomic amplification of RTK genes, resulting in the increase of local concentrations of RTKs.
3. Epidermal Growth Factor Receptors
A common family of RTKs often observed as overexpressed in ovarian cancer is the epidermal growth factor receptor (EGFR) or ErbB family. Consisting of EGFR (also referred to as HER1/ErbB1), HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4), this family of structurally related receptors are essential in normal cellular development, proliferation, differentiation, and survival [34]. When the EGFR is abnormally activated, the dimerization or over-expression of ligand-dependent receptors leads to the manifestation of tumours which are epithelial in origin [35]. The ErbB receptor signalling occurs via MAPK, STAT, PI3K, and mTOR pathways, important for cell survival, proliferation, and differentiation, assisting in the oncogenic activity of the ErbB family [36]. The number of OC with EGFR activating mutations and amplification are small (<4% and 4–22%, respectively) compared to other cancers such as lung cancers [37]. EGFR overexpression, on the other hand, varies from 9–62%, depending on assay conditions and selected cut-off values [37]. While EGFR is commonly referenced in the literature, there is limited knowledge regarding associations between EGFR expression and disease outcome [38].
The association between HER2 expression and OC prognosis has been investigated, with HER2 amplification being non-prognostic [39]. A meta-analysis of 34 studies involving 5180 OC patients showed that expression of HER2 was negatively correlated with overall survival (OS) [40]. More recent data studying HER2 and HER3 in the same cohort of 105 cases suggest 3.8% positivity of HER2 by immunohistochemistry and 5.7% by in situ hybridization. In contrast, HER3 levels were higher, being 12.4% and 8.6%, respectively. Their study did not identify significant correlation between HER2 status and survival. However, the HER3 status by fluorescence in situ hybridization was associated with poor progression-free survival (PFS) [41].
4. Vascular Endothelial Growth Factor Receptor and Its Pathways
A study has used 339 primary ovarian tumours to show that vascular endothelial growth factor (VEGF) was overexpressed in only 7% of tumours and was correlated with significantly poorer survival [42]. This suggests that the therapeutic success of targeting the VEGF pathway in isolation is limited to only a small subset of patients.
Subsequent studies that examined VEGF receptors and their ligands compared primary high grade serous ovarian tumours and their paired distant omental metastases. The study showed that the protein expressions of VEGF-A, VEGF-D, and VEGFR1 were higher in the metastases than the primary lesions [43]. Significant indicators associated with short PFS include high VEGF-C level, low VEGFR3, and low epithelial expression of VEGF-A. The reasoning for these associations were not clear, although the survival analysis was univariate.
5. Platelet-Derived Growth Factor Receptors
Platelet-derived growth factor (PDGF) and PDGFR-α expression were mostly detected in the malignant ovarian tumours as compared to the benign tumours and the normal ovaries through immunohistochemistry. PDGFR-ß was not detected in either the normal epithelium or the tumour cells. Patients with PDGFR-α positive tumour cells were associated with shorter survival compared to negative tumours, suggesting the prognostic role of PGDFR-α in OC [44]. More recent investigation suggested that PDGFR-α and PDGFR-ß were not related with FIGO stage, grade, or histopathological subtype of OCs; but PDGFR-ß expression in cancer cells was associated with improved OS in 52 cases. The significance of this correlation in relation to therapy design is still yet to be fully understood [45].
6. c-MET
A meta-analysis study on c-MET in OCs involving 568 patients from seven studies suggested that patients with high levels of c-MET in tumours were associated with worse survival than those with low tumour c-MET level [46]. Although not statistically significant, there was a trend of c-MET overexpression associated with higher FIGO stage and lymph node metastases. Hence, c-MET expression may serve as a prognostic marker and may be useful to target in OC [46]. Other studies concluded that c-MET expression was greater in OCCC than the serous carcinoma subtype [47].
7. Other Emerging RTKs
A key RTK family influencing the cellular behaviour of OC is the Axl family; however, the inhibition of Axl as treatment has mainly been explored preclinically [48]. This family consists of three RTKs, namely Tyro3, Axl, and MerTK (or TAM receptors) [49]. Within the literature there are several alternate names for each receptor. Tyro3 is also referred to as c-Eyk, c-mer, MER, RP38, and Tryo12. Axl can also be called ARK, JTK11, Tyro7, and UFO. Tyro3 is also known as BYK, Etk-2, DTK, Rek, RSE, Sky, and Tif. Axl overexpression has been detected in multiple cancers, known to have a supportive role in tumorigenesis [50]. In chemoresistant OC cell samples, Axl has been observed to be expressed in high levels [48]. Ligand activated Axl promotes proliferation in tumours and its overexpression is correlated with poor prognosis in patients with OC [48]. Various studies have demonstrated the correlation between chemoresistance in OC cells whereby the inhibition of Axl resulted in an increased chemosensitivity of the HGSC cells to platinum-based therapeutics [48][51]. In the same RTK family, Tyro3 was found to be upregulated in Taxol-resistant cell lines, with a reported role of promoting proliferation in OC cells [52]. The inhibition of Tyro3 produced a reversal in the chemoresistance cell lines, restoring sensitivity to Taxol [49][53]
Another RTK target with promising preclinical results and potential influence over chemoresistance in OC is the receptor tyrosine kinase-like orphan receptor (ROR) family. Recent studies have demonstrated trends of elevated ROR1 and ROR2 in chemoresistant HGSC cell lines [54][55]. Forming part of the Wnt signalling pathway, the receptors have an important role in epithelial-mesenchymal transition, alluding to potential metastatic and chemoresistant influences [56]. Studies conducted by Henry et al., further established a strong correlation between these orphan receptors, with combined inhibition of both ROR1 and ROR2 demonstrating a significant chemo-sensitising effect to cisplatin in OC cell lines [54][55]. Whilst further study will be required to dissect the exact mechanism of each receptor in isolation, the ROR family represents a promising new target for potential therapeutic interventions.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10092113
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Prat, J. FIGO’s staging classification for cancer of the ovary, fallopian tube, and peritoneum: Abridged republication. J. Gynecol. Oncol. 2015, 26, 87–89.
- Dilley, J.; Burnell, M.; Gentry-Maharaj, A.; Ryan, A.; Neophytou, C.; Apostolidou, S.; Karpinskyj, C.; Kalsi, J.; Mould, T.; Woolas, R.; et al. Ovarian cancer symptoms, routes to diagnosis and survival–Population cohort study in the ‘no screen’arm of the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS). Gynecol. Oncol. 2020, 158, 316–322.
- Gajjar, K.; Ogden, G.; Mujahid, M.; Razvi, K. Symptoms and risk factors of ovarian cancer: A survey in primary care. Int. Sch. Res. Not. 2012, 2012, 754197.
- Berek, J.S.; Bast, R.C., Jr. Epithelial ovarian cancer. In Holland-Frei Cancer Medicine, 6th ed.; BC Decker: Hamilton, ON, Canada, 2003.
- Kobel, M.; Kalloger, S.E.; Boyd, N.; McKinney, S.; Mehl, E.; Palmer, C.; Leung, S.; Bowen, N.J.; Lonescu, D.N.; Rajput, A.; et al. Ovarian carcinoma subtypes are different diseases: Implications for biomarker studies. PLoS Med. 2008, 5, 232.
- Zhou, J.; Wu, S.-G.; Wang, J.; Sun, J.-Y.; He, Z.-Y.; Jin, X.; Zhang, W.-W. The effect of histological subtypes on outcomes of stage IV epithelial ovarian cancer. Front. Oncol. 2018, 8, 577.
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian cancers: Genetic abnormalities, tumor heterogeneity and progression, clonal evolution and cancer stem cells. Medicines 2018, 5, 16.
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56.
- Ryland, G.L.; Hunter, S.M.; Doyle, M.A.; Caramia, F.; Li, J.; Rowley, S.M.; Christie, M.; Allan, P.E.; Stephens, A.N.; Bowtell, D.D.L.; et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Med. 2015, 7, 1–12.
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615.
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.-W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106, dju249.
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208.
- Chen, G.M.; Kannan, L.; Geistlinger, L.; Kofia, V.; Safikhani, Z.; Gendoo, D.M.A.; Parmigiani, G.; Birrer, M.; Haibe-Kains, B.; Waldron, L. Consensus on Molecular Subtypes of High-Grade Serous Ovarian CarcinomaConsensus on Molecular Subtypes of HGSOC. Clin. Cancer Res. 2018, 24, 5037–5047.
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38.
- Altman, A.D.; Nelson, G.; Chu, P.; Nation, J.; Ghatage, P. Optimal debulking targets in women with advanced stage ovarian cancer: A retrospective study of immediate versus interval debulking surgery. J. Obstetr. Gynaecol. Can. 2012, 34, 558–566.
- Eisenkop, S.M.; Spirtos, N.M. What are the current surgical objectives, strategies, and technical capabilities of gynecologic oncologists treating advanced epithelial ovarian cancer? Gynecol. Oncol. 2001, 82, 489–497.
- Tewari, D.; Java, J.J.; Salani, R.; Armstrong, D.K.; Markman, M.; Herzog, T.; Monk, B.J.; Chan, J.K. Long-term survival advantage and prognostic factors associated with intraperitoneal chemotherapy treatment in advanced ovarian cancer: A gynecologic oncology group study. J. Clin. Oncol. 2015, 33, 1460–1466.
- Christie, E.L.; Bowtell, D.D.L. Acquired chemotherapy resistance in ovarian cancer. Ann. Oncol. 2017, 28, viii13–viii15.
- Cannistra, S.A. Cancer of the ovary. N. Engl. J. Med. 2004, 351, 2519–2529.
- National Institutes of Health Consensus Development Conference Statement. Ovarian cancer: Screening, treatment, and follow-up. Gynecol. Oncol. 1994, 55, S4–S14.
- Ushijima, K. Treatment for recurrent ovarian cancer—At first relapse. J. Oncol. 2010, 2010, 497429.
- Klempner, S.J.; Myers, A.P.; Mills, G.B.; Westin, S.N. Clinical investigation of receptor and non-receptor tyrosine kinase inhibitors for the treatment of epithelial ovarian cancer. Expert Opin. Pharmacother. 2013, 14, 2171–2182.
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134.
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1659–1672.
- Le Tourneau, C.; Raymond, E.; Faivre, S. Sunitinib: A novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST). Ther. Clin. Risk Manag. 2007, 3, 341–348.
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58.
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase–role and significance in cancer. Int. J. Med. Sci. 2004, 1, 101–115.
- Mayer, B.J. Perspective: Dynamics of receptor tyrosine kinase signaling complexes. FEBS Lett. 2012, 586, 2575–2579.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558.
- Metibemu, D.S.; Akinloye, O.A.; Akamo, A.J.; Ojo, D.A.; Okeowo, O.T.; Omotuyi, I.O. Exploring receptor tyrosine kinases-inhibitors in Cancer treatments. Egypt J. Med. Hum. Genet. 2019, 20, 35.
- Xu, A.M.; Huang, P.H. Receptor Tyrosine Kinase Coactivation Networks in CancerRTK Coactivation Networks. Cancer Res. 2010, 70, 3857–3860.
- Jiao, Y.; Ou, W.; Meng, F.; Zhou, H.; Wang, A. Targeting HSP90 in ovarian cancers with multiple receptor tyrosine kinase coactivation. Mol. Cancer 2011, 10, 125.
- Tvorogov, D.; Carpenter, G. Epidermal Growth Factor Receptor Family. In Encyclopedia of Biological Chemistry; Lennarz, W.J., Lane, M.D., Eds.; Elsevier: New York, NY, USA, 2004; pp. 51–55.
- Cao, C.; Lu, S.; Sowa, A.; Kivlin, R.; Amaral, A.; Chu, W.; Yang, H.; Di, W.; Wen, Y. Priming with EGFR tyrosine kinase inhibitor and EGF sensitizes ovarian cancer cells to respond to chemotherapeutical drugs. Cancer Lett. 2008, 266, 249–262.
- Teplinsky, E.; Muggia, F. EGFR and HER2: Is there a role in ovarian cancer? Transl. Cancer Res. 2015, 4, 1.
- Sheng, Q.; Liu, J. The therapeutic potential of targeting the EGFR family in epithelial ovarian cancer. Br. J. Cancer 2011, 104, 1241–1245.
- Mehner, C.; Oberg, A.L.; Goergen, K.M.; Kalli, K.R.; Maurer, M.J.; Nassar, A.; Goode, E.L.; Keeney, G.L.; Jatoi, A.; Radisky, D.C. EGFR as a prognostic biomarker and therapeutic target in ovarian cancer: Evaluation of patient cohort and literature review. Genes Cancer 2017, 8, 589.
- McAlpine, J.N.; Wiegand, K.C.; Vang, R.; Ronnett, B.M.; Adamiak, A.; Köbel, M.; Kalloger, S.E.; Swenerton, K.D.; Huntsman, D.G.; Gilks, C.B.; et al. HER2 overexpression and amplification is present in a subset of ovarian mucinous carcinomas and can be targeted with trastuzumab therapy. BMC Cancer 2009, 9, 433.
- Luo, H.; Xu, X.; Ye, M.; Sheng, B.; Zhu, X. The prognostic value of HER2 in ovarian cancer: A meta-analysis of observational studies. PLoS ONE 2018, 13, 0191972.
- Chung, Y.W.; Kim, S.; Hong, J.H.; Lee, J.K.; Lee, N.W.; Lee, Y.S.; Song, J.Y. Overexpression of HER2/HER3 and clinical feature of ovarian cancer. J. Gynecol. Oncol. 2019, 30, e75.
- Duncan, T.J.; Al-Attar, A.; Rolland, P.; Scott, I.V.; Deen, S.; Liu, D.T.; Spendlove, I.; Durrant, L.G. Vascular endothelial growth factor expression in ovarian cancer: A model for targeted use of novel therapies? Clin. Cancer Res. 2008, 14, 3030–3035.
- Sopo, M.; Anttila, M.; Hämäläinen, K.; Kivelä, A.; Ylä-Herttuala, S.; Kosma, V.-M.; Keski-Nisula, L.; Sallinen, H. Expression profiles of VEGF-A, VEGF-D and VEGFR1 are higher in distant metastases than in matched primary high grade epithelial ovarian cancer. BMC Cancer 2019, 19, 584.
- Henriksen, R.; Funa, K.; Wilander, E.; Bäckström, T.; Ridderheim, M.; Öberg, K. Expression and prognostic significance of platelet-derived growth factor and its receptors in epithelial ovarian neoplasms. Cancer Res. 1993, 53, 4550–4554.
- Szubert, S.; Moszynski, R.; Szpurek, D.; Romaniuk, B.; Sajdak, S.; Nowicki, M.; Michalak, S. The expression of Platelet-derived Growth factor receptors (PDGFRs) and their correlation with overall survival of patients with ovarian cancer. Ginekol. Pol. 2019, 90, 242–249.
- Kim, J.H.; Jang, H.J.; Kim, H.S.; Kim, B.J.; Park, S.H. Prognostic impact of high c-Met expression in ovarian cancer: A meta-analysis. J. Cancer 2018, 9, 3427–3434.
- Kim, H.-J.; Yoon, A.; Ryu, J.-Y.; Cho, Y.-J.; Choi, J.-J.; Song, S.Y.; Bang, H.; Lee, J.S.; Cho, W.C.; Choi, C.H. c-MET as a potential therapeutic target in ovarian clear cell carcinoma. Sci. Rep. 2016, 6, 38502.
- Quinn, J.M.; Greenwade, M.M.; Palisoul, M.L.; Opara, G.; Massad, K.; Guo, L.; Zhao, P.; Beck-Noia, H.; Hagemann, I.S.; Hangemann, A.R. Therapeutic Inhibition of the Receptor Tyrosine Kinase AXL Improves Sensitivity to Platinum and Taxane in Ovarian CancerAXL Inhibition Improves Chemosensitivity in Ovarian Cancer. Mol. Cancer Ther. 2019, 18, 389–398.
- Vouri, M.; Hafizi, S. TAM receptor tyrosine kinases in cancer drug resistance. Cancer Res. 2017, 77, 2775–2778.
- Rankin, E.B.; Giaccia, A.J. The receptor tyrosine kinase AXL in cancer progression. Cancers 2016, 8, 103.
- Tian, M.; Chen, X.S.; Li, L.Y.; Wu, H.Z.; Zeng, D.; Wang, X.L.; Zhang, Y.; Xioa, S.-S.; Cheng, Y. Inhibition of AXL enhances chemosensitivity of human ovarian cancer cells to cisplatin via decreasing glycolysis. Acta Pharmacol. Sin. 2021, 42, 1180–1189.
- Lee, C. Overexpression of Tyro3 receptor tyrosine kinase leads to the acquisition of taxol resistance in ovarian cancer cells. Mol. Med. Rep. 2015, 12, 1485–1492.
- Suh, Y.A.; Jo, S.Y.; Lee, H.Y.; Lee, C. Inhibition of IL-6/STAT3 axis and targeting Axl and Tyro3 receptor tyrosine kinases by apigenin circumvent taxol resistance in ovarian cancer cells. Int. J. Oncol. 2015, 46, 1405–1411.
- Henry, C.; Llamosas, E.; Knipprath-Meszaros, A.; Schoetzau, A.; Obermann, E.; Fuenfschilling, M.; Caduff, R.; Fink, D.; Hacker, N.; Ward, R.; et al. Targeting the ROR1 and ROR2 receptors in epithelial ovarian cancer inhibits cell migration and invasion. Oncotarget 2015, 6, 40310–40326.
- Henry, C.; Llamosas, E.; Djordjevic, A.; Hacker, N.; Ford, C. Migration and invasion is inhibited by silencing ROR1 and ROR2 in chemoresistant ovarian cancer. Oncogenesis 2016, 5, e226.
- Hossein, G.; Arabzadeh, S.; Salehi-Dulabi, Z.; Dehghani-Ghobadi, Z.; Heidarian, Y.; Talebi-Juybari, M. Wnt5A regulates the expression of ROR2 tyrosine kinase receptor in ovarian cancer cells. Biochem. Cell Biol. 2017, 95, 609–615.
This entry is offline, you can click here to edit this entry!