The sideroblastic anemias are a heterogeneous group of inherited and acquired disorders characterized by anemia and the presence of ring sideroblasts in the bone marrow. Ring sideroblasts are abnormal erythroblasts with iron-loaded mitochondria that are visualized by Prussian blue staining as a perinuclear ring of green-blue granules. The mechanisms that lead to the ring sideroblast formation are heterogeneous, but in all of them, there is an abnormal deposition of iron in the mitochondria of erythroblasts. Congenital sideroblastic anemias include nonsyndromic and syndromic disorders. Acquired sideroblastic anemias include conditions that range from clonal disorders (myeloid neoplasms as myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms with ring sideroblasts) to toxic or metabolic reversible sideroblastic anemia. Due to the advances in genomic techniques, a deep knowledge of the pathophysiological mechanisms has been accomplished and the bases for possible targeted treatments have been established. The distinction between the different forms of sideroblastic anemia is based on the study of the characteristics of the anemia, age of diagnosis, clinical manifestations, and the performance of laboratory analysis involving genetic testing in many cases.
- sideroblastic anemia
- ring sideroblast
- MDS
- MDS/MPN-RS-T
- SF3B1
1. Introduction
2. Historical Context
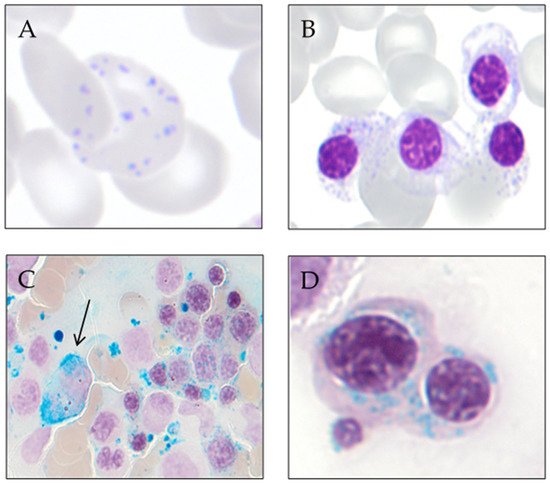
3. Ring Sideroblast Definition
- -
-
Type 1 sideroblasts: < 5 siderotic granules in the cytoplasm.
- -
-
Type 2 sideroblasts: ≥ 5 siderotic granules, but no perinuclear distribution.
- -
-
Type 3 or ring sideroblasts: ≥ 5 siderotic granules in a perinuclear position, covering at least one-third of the nuclear circumference.
4. Ring Sideroblast Formation
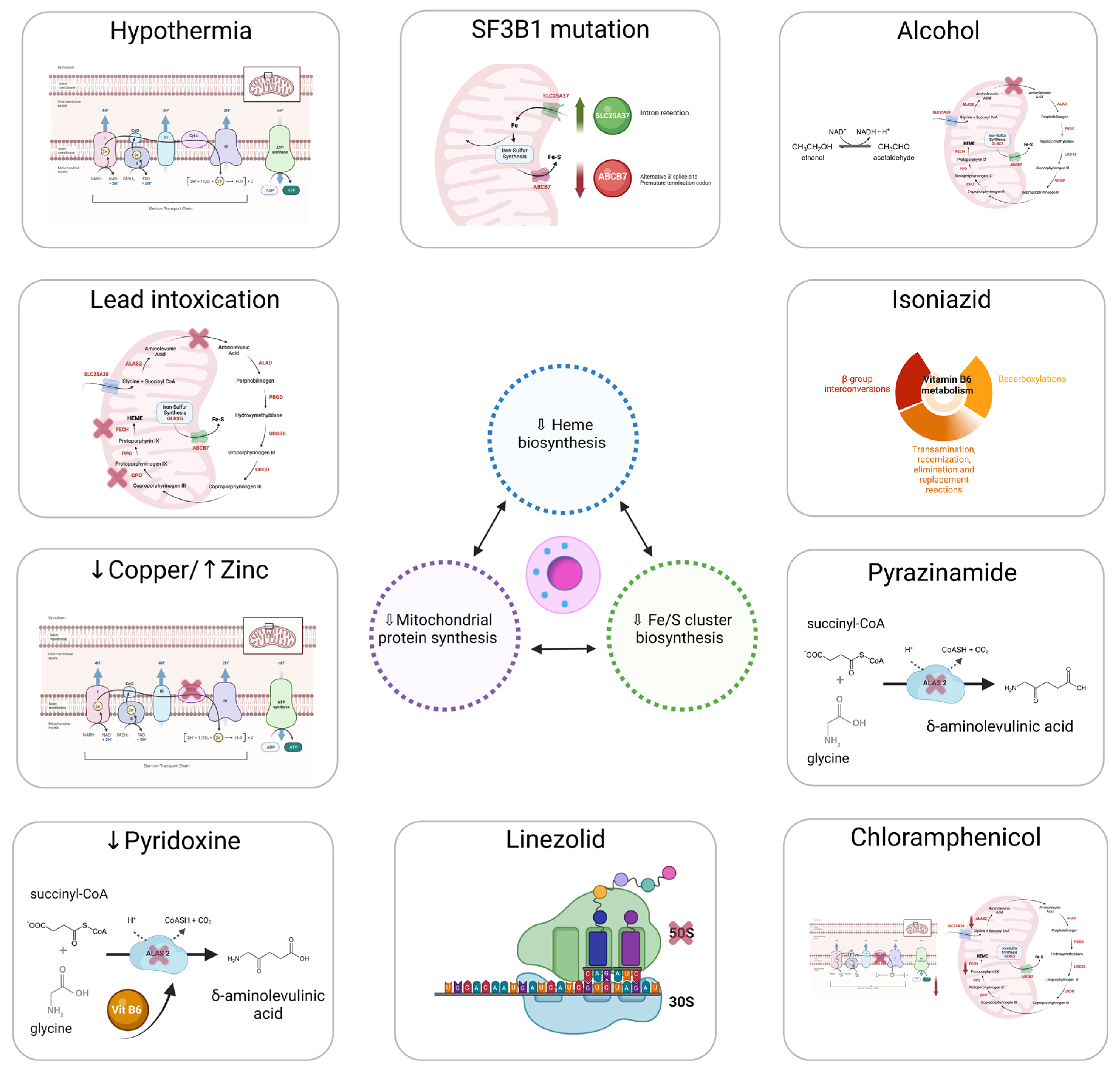
4.1. Heme Synthesis
4.2. ISC Biogenesis
4.3. Mitochondrial Respiratory Complex Proteins and Mitochondrial Protein Synthesis
5. Diagnosis of Sideroblastic Anemias
This entry is adapted from the peer-reviewed paper 10.3390/genes13091562
References
- Ducamp, S.; Fleming, M.D. The molecular genetics of sideroblastic anemia. Blood 2019, 133, 59–69.
- Grüneberg, H. Siderocytes: A New Kind of Erythrocytes. Nature 1941, 148, 114–115.
- Cooley, T. A severe type of hereditary anemia with elliptocytosis. Interesting sequences of splenectomy. Am. J. Med. Sci. 1945, 209, 561–568.
- Cotter, P.D.; Rucknagel, D.L.; Bishop, D.F. X-linked sideroblastic anemia: Identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene (ALAS2) in the original family described by Cooley. Blood 1994, 84, 3915–3924.
- Bjorkman, S.E. Chronic refractory anemia with sideroblastic bone marrow; a study of four cases. Blood 1956, 11, 250–259.
- Mollin, D.L. A symposium on sideroblastic anaemia held at the Postgraduate Medical School of London on Friday, March 20th, 1964, during the Annual Meeting of the British Society for Haematology. Introduction: Sideroblasts and Sideroblastic Anaemia. Br. J. Haematol. 1965, 11, 41–48.
- Swerdlow, S.H.; World Health Organization. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2017.
- Dharwadkar, A.; Vimal, S.; Panicker, N.K.; Chandanwale, S.S.; Viswanathan, V.; Kumar, H. Study of sideroblasts and iron stores in bone marrow aspirates using Perls′ stain. Med. J. Dr. DY Patil Univ. 2016, 9, 181.
- Juneja, S.K.; Imbert, M.; Jouault, H.; Scoazec, J.Y.; Sigaux, F.; Sultan, C. Haematological features of primary myelodysplastic syndromes (PMDS) at initial presentation: A study of 118 cases. J. Clin. Pathol. 1983, 36, 1129–1135.
- Mufti, G.J.; Bennett, J.M.; Goasguen, J.; Bain, B.J.; Baumann, I.; Brunning, R.; Cazzola, M.; Fenaux, P.; Germing, U.; Hellström-Lindberg, E.; et al. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of myelodysplastic syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica 2008, 93, 1712–1717.
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2008; Volume 2.
- Abu-Zeinah, G.; DeSancho, M.T. Understanding Sideroblastic Anemia: An Overview of Genetics, Epidemiology, Pathophysiology and Current Therapeutic Options. J. Blood Med. 2020, 11, 305–318.
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002.
- Minakami, S.; Yoshikawa, H. Studies on erythrocyte glycolysis. II. Free energy changes and rate limitings steps in erythrocyte glycolysis. J. Biochem. 1966, 59, 139–144.
- Jansen, R.P. Origin and persistence of the mitochondrial genome. Hum. Reprod. 2000, 15, 1–10.
- Zardoya, R. Recent advances in understanding mitochondrial genome diversity. F1000Research 2020, 9, 270.
- McCormick, E.M.; Muraresku, C.C.; Falk, M.J. Mitochondrial Genomics: A Complex Field Now Coming of Age. Curr. Genet. Med. Rep. 2018, 6, 52–61.
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780.
- Sulaiman, S.A.; Rani, Z.M.; Radin, F.Z.M.; Murad, N.A.A. Advancement in the diagnosis of mitochondrial diseases. J. Transl. Genet. Genom. 2020, 4, 159–187.
- Kuroiwa, T. The discovery of the division apparatus of plastids and mitochondria. QJM Int. J. Med. 2000, 49, 123–134.
- Broker, S.; Meunier, B.; Rich, P.; Gattermann, N.; Hofhaus, G. MtDNA mutations associated with sideroblastic anaemia cause a defect of mitochondrial cytochrome c oxidase. JBIC J. Biol. Inorg. Chem. 1998, 258, 132–138.
- Gattermann, N.; Retzlaff, S.; Wang, Y.L.; Hofhaus, G.; Heinisch, J.; Aul, C.; Schneider, W. Heteroplasmic point mutations of mitochondrial DNA affecting subunit I of cytochrome c oxidase in two patients with acquired idiopathic sideroblastic anemia. Blood 1997, 90, 4961–4972.
- Rascati, R.; Parsons, P. Purification and characterization of cytochrome c oxidase from rat liver mitochondria. J. Biol. Chem. 1979, 254, 1586–1593.
- Chen, J.-J. Regulation of protein synthesis by the heme-regulated eIF2α kinase: Relevance to anemias. Blood 2006, 109, 2693–2699.
- Bottomley, S.S.; Muller-Eberhard, U. Pathophysiology of heme synthesis. Semin. Hematol. 1988, 25, 282–302.
- Fujiwara, T.; Harigae, H. Biology of Heme in Mammalian Erythroid Cells and Related Disorders. BioMed Res. Int. 2015, 2015, 278536.
- Rudd, D.M. Elsevier’s Integrated Review Biochemistry, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2012.
- Ponka, P. Tissue-Specific Regulation of Iron Metabolism and Heme Synthesis: Distinct Control Mechanisms in Erythroid. Cells Blood 1997, 89, 1–25.
- Mustajoki, S.; Laine, M.; Lahtela, M.; Mustajoki, P.; Peltonen, L.; Kauppinen, R. Acute Intermittent Porphyria: Expression of Mutant and Wild-Type Porphobilinogen Deaminase in COS-1 Cells. Mol. Med. 2000, 6, 670–679.
- Rademakers, L.H.P.M.; Koningsberger, J.C.; Sorber, C.W.J.; DE LA Faille, H.B.; VAN Hattum, J.; Marx, J.J.M. Accumulation of iron in erythroblasts of patients with erythropoietic protoporphyria. Eur. J. Clin. Investig. 1993, 23, 130–138.
- Lecha, M.; Puy, H.; Deybach, J.C. Erythropoietic protoporphyria. Orphanet. J. Rare Dis. 2009, 4, 19.
- Bishop, D.F.; Henderson, A.S.; Astrin, K.H. Human delta-aminolevulinate synthase: Assignment of the housekeeping gene to 3p21 and the erythroid-specific gene to the X chromosome. Genomics 1990, 7, 207–214.
- Riddle, R.D.; Yamamoto, M.; Engel, J.D. Expression of delta-aminolevulinate synthase in avian cells: Separate genes encode erythroid-specific and nonspecific isozymes. Proc. Natl. Acad. Sci. USA 1989, 86, 792–796.
- Cotter, P.D.; Willard, H.F.; Gorski, J.L.; Bishop, D.F. Assignment of human erythroid delta-aminolevulinate synthase (ALAS2) to a distal subregion of band Xp11.21 by PCR analysis of somatic cell hybrids containing X; autosome translocations. Genomics 1992, 13, 211–212.
- Cox, T.C.; Bawden, M.J.; Martin, A.; May, B.K. Human erythroid 5-aminolevulinate synthase: Promoter analysis and identification of an iron-responsive element in the mRNA. EMBO J. 1991, 10, 1891–1902.
- Bergmann, A.K.; Bs, D.R.C.; BS, E.M.M.; Agarwal, S.; Fleming, M.D.; Bottomley, S.S.; Neufeld, E.J. Systematic molecular genetic analysis of congenital sideroblastic anemia: Evidence for genetic heterogeneity and identification of novel mutations. Pediatr. Blood Cancer 2009, 54, 273–278.
- Stehling, O.; Lill, R. The Role of Mitochondria in Cellular Iron-Sulfur Protein Biogenesis: Mechanisms, Connected Processes, and Diseases. Cold Spring Harb. Perspect. Biol. 2013, 5, a011312.
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemias: Molecular basis, pathophysiology, and clinical aspects. In Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine; Porphyrias and Sideroblastic Anemias, World Scientific: Singapore, 2014; Volume 29, pp. 43–87.
- Zhang, D.-L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124.
- Hamdi, A.; Roshan, T.M.; Kahawita, T.M.; Mason, A.B.; Sheftel, A.D.; Ponka, P. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim. Biophys. Acta 2016, 1863, 2859–2867.
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272.
- Drysdale, J.; Arosio, P.; Invernizzi, R.; Cazzola, M.; Volz, A.; Corsi, B.; Biasiotto, G.; Levi, S. Mitochondrial Ferritin: A New Player in Iron Metabolism. Blood Cells Mol. Dis. 2002, 29, 376–383.
- Bou-Abdallah, F.; Santambrogio, P.; Levi, S.; Arosio, P.; Chasteen, N.D. Unique Iron Binding and Oxidation Properties of Human Mitochondrial Ferritin: A Comparative Analysis with Human H-chain Ferritin. J. Mol. Biol. 2005, 347, 543–554.
- Nie, G.; Sheftel, A.D.; Kim, S.F.; Ponka, P.; Brown, P.; Levis, M.; Shurtleff, S.; Campana, D.; Downing, J.; Small, D. Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood 2005, 105, 2161–2167.
- Bessis, M.C.; Breton-Gorius, J. Ferritin and Ferruginous Micelles in Normal Erythroblasts and Hypochromic Hypersideremic Anemias. Blood 1959, 14, 423–432.
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A Human Mitochondrial Ferritin Encoded by an Intronless Gene. J. Biol. Chem. 2001, 276, 24437–24440.
- Cazzola, M.; Invernizzi, R.; Bergamaschi, G.; Levi, S.; Corsi, B.; Travaglino, E.; Rolandi, V.; Biasiotto, G.; Drysdale, J.; Arosio, P. Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood 2003, 101, 1996–2000.
- Liochev, S.I.; Fridovich, I. The role of O2.- in the production of HO.: In vitro and in vivo. Free Radic. Biol. Med. 1994, 16, 29–33.
- Gutteridge, J.M.; Rowley, D.A.; Halliwell, B. Superoxide-dependent formation of hydroxyl radicals in the presence of iron salts. Detection of ‘free’ iron in biological systems by using bleomycin-dependent degradation of DNA. Biochem. J. 1981, 199, 263–265.
- Xu, W.; Barrientos, T.; Andrews, N.C. Iron and Copper in Mitochondrial Diseases. Cell Metab. 2013, 17, 319–328.
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Oncol. Clin. N. Am. 2014, 28, 653–670.
- Camaschella, C. Hereditary Sideroblastic Anemias: Pathophysiology, Diagnosis, and Treatment. Semin. Hematol. 2009, 46, 371–377.
- Tanno, T.; Miller, J.L. Iron Loading and Overloading due to Ineffective Erythropoiesis. Adv. Hematol. 2010, 2010, 358283.
- Ohba, R.; Furuyama, K.; Yoshida, K.; Fujiwara, T.; Fukuhara, N.; Onishi, Y.; Manabe, A.; Ito, E.; Ozawa, K.; Kojima, S.; et al. Clinical and genetic characteristics of congenital sideroblastic anemia: Comparison with myelodysplastic syndrome with ring sideroblast (MDS-RS). Ann. Hematol. 2012, 92, 1–9.
- Zahid, M.F.; Khan, N.; Pei, J.; Testa, J.R.; Dulaimi, E. Genomic imbalances in peripheral blood confirm the diagnosis of myelodysplastic syndrome in a patient presenting with non-immune hemolytic anemia. Leuk. Res. Rep. 2016, 5, 23–26.
- Woessner, S.F.L. La Citología Óptica en el Diagnóstico Hematológico, 5th ed.; Acción Médica: Madrid, Spain, 2006.
- Acín, P.; Florensa, L.; Andreu, L.; Woessner, S. Cytoplasmic abnormalities of erythroblasts as a marker for ringed sideroblasts in myelodysplastic syndromes. Eur. J. Haematol. 2009, 54, 276–278.
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984.