Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
The World Health Organization announced that COVID-19, with SARS-CoV-2 as its pathogen, had become a pandemic on 11 March 2020. With the development of research, cardiovascular injury in patients with COVID-19, such as arrhythmia, myocardial injury, and heart failure, is the second major symptom in addition to respiratory symptoms, and cardiovascular injury is related to the prognosis and mortality of patients. The incidence of arrhythmia in COVID-19 patients ranges from 10% to 20%.
- COVID-19
- SARS-CoV-2
- arrhythmia
1. Introduction
On 31 December 2019, severe acute respiratory syndrome caused by a novel coronavirus was officially reported for the first time in Wuhan, Hubei Province, China [1]. The new disease spread rapidly at an astonishing rate. As of 5 March 2022, there were nearly 600 million cumulative cases and nearly 6.5 million deaths worldwide, and these data are still increasing [2]. The pathogen of the disease was identified by the World Health Organization and named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
The most common symptoms of people infected with SARS-CoV-2 are cough, fever and fatigue, and there are many asymptomatic infections. The most common complications in COVID-19 were acute respiratory distress syndrome (RDS), arrhythmia and shock, accounting for 25% to 35% of all complications [3,4,5]. Older people with underlying diseases such as hypertension, liver, kidney, and cardiovascular diseases have higher mortality rates [5,6]. There is increasing evidence that mortality rate in patients with COVID-19 is associated with cardiovascular comorbidities, and some possible mechanisms have been proposed.
2. Incidence and Clinical Manifestations of COVID-19 Complicated with Arrhythmia
2.1. Incidence of Disease
In the past, severe acute respiratory syndrome virus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS) were also coronaviruses. In the coronavirus pandemics caused by SARS-CoV and MERS, myocardial infarction, acute myocarditis, heart failure and other cardiac manifestations were reported [5]. Therefore, researchers speculated that these complications may also be associated with COVID-19 [5]. In fact, several retrospective studies have confirmed a correlation between cardiovascular risk factors and the prognosis and mortality of COVID-19. One of the most important risk factors for cardiovascular diseases in patients with COVID-19 is cardiovascular disease that exists before infection [7].
In the largest available worldwide survey [8], which involves 4526 hospitalized COVID-19 patients throughout the world, 18.27% of patients developed arrhythmias because of COVID-19. In these patients, 81.8% developed atrial arrhythmias, 20.7% developed ventricular arrhythmias, and the vast majority were tachyarrhythmias and only 22.6% were bradyarrhythmias. The results were not very different from other small and medium-sized studies in the past, where the incidence of arrhythmia in COVID-19 patients ranges from 10% to 20%. In a retrospective study in Hubei, China, nearly 7.3% of people reported palpitation as the first symptom [9]. The study of Zhao et al. [10] showed that myocardial injury is the most common cardiovascular complications in patients with COVID-19 (21.2%), followed by arrhythmia (15.3%), heart failure (14.4%) and acute coronary syndrome (1.0%). In the death cases, the combined incidence of arrhythmia, heart failure and myocardial injury was 47.8%, 40.3% and 61.7%, respectively. Sahranavard et al.’s analysis of 22 studies in 4157 patients found that the incidence of arrhythmias was 10.11% [11]. In a study involving 138 patients with COVID-19, arrhythmias accounted for 19.6% of all complications, and the incidence of ventricular tachycardia or ventricular fibrillation (malignant ventricular arrhythmias) was 5.9% [12]. At the same time, several studies have found that the incidence of arrhythmias in critically ill patients is higher than that in noncritical patients [13]. The incidence of arrhythmia in ICU patients was 37%, of which 68% were new [14], similar to the results of Bhatia et al. (43%) [15].
2.2. Clinical Manifestation
There are many manifestations of arrhythmia in patients with COVID-19. Most studies did not record the specific causes and types of such arrhythmias, but relevant records and data are constantly emerging. For example, in a retrospective cohort study involving 393 cases in the United States, 7.1% of patients had atrial arrhythmia [16], while in another study involving 115 cases, 16.5% of patients had atrial arrhythmia [17]. A series of cases of arrhythmia caused by SARS-CoV-2 infection showed high-grade atrioventricular block, new atrial fibrillation, long-short sequence polymorphic ventricular tachycardia caused by ventricular premature beats, and cardiac arrest with pulseless electrical activity [18]. A retrospective study of 187 patients [19] found that mortality in COVID-19 patients was associated with cerebrovascular diseases and myocardial injury, and 5.9% of these patients had ventricular tachycardia or ventricular fibrillation.
Studies have shown that the most common tachyarrhythmia in COVID-19 patients is atrial fibrillation [20], and the most common bradyarrhythmias are severe sinus bradycardia and complete heart block [20]. Tachyarrhythmia such as atrial flutter, ventricular tachycardia, atrial fibrillation, and supraventricular tachycardia has been reported in some literature. Multiple meta-analyses and studies have found that the incidence of atrial fibrillation in COVID-19 patients was around 8–13% [21,22,23], particularly common in elderly and seriously ill patients [22]. This is similar to the incidence of atrial fibrillation in influenza patients (12%) [23], so it can be speculated that atrial fibrillation is not specific to COVID-19 patients and that it is probably a common complication of the systemic inflammatory response in viral infection.
In addition, the probability of supraventricular tachyarrhythmia in severe COVID-19 patients was high, and atrial fibrillation may be a sign of critical illness, which was the most common arrhythmia in critical nursing environments [18,20].
A meta-analysis [24] that included 28 studies showed that the most common arrhythmias were supraventricular arrhythmias (6.2%) and ventricular arrhythmias (2.5%). Their study also noted that COVID-19 patients were prone to exhibit ECG features such as QTc prolongation (12.3%) and ST-segment deviation (8.7%), but the prognostic significance of these features is unclear [24]. This study result is like a retrospective study that counted data from COVID-19 patients with concomitant arrhythmias worldwide, which noted that 81.8% of those who developed arrhythmias has atrial arrhythmias, including atrial fibrillation, atrial flutter and supraventricular tachycardia [8].
Meanwhile, although tachyarrhythmias are the most common, 22.6% of patients may still develop bradyarrhythmias, such as bradycardia and atrioventricular block [8]. In the study by Lao et al. [25], the incidence of new-onset atrioventricular block in COVID-19 patients was 5.5%, most of which were benign. However, the study by Qingxing Chen et al. [26] concluded that ventricular tachycardia and atrioventricular block are uncommon and are most often seen at the end stage of disease in critically ill patients, and they suggested that these types of arrhythmias may serve as a sign of disease progression. For instance, a male with COVID-19 symptoms showed atrioventricular block at 2:1 and was subsequently confirmed to be infected [27]. Bradycardia has also been reported in some literature. One case of transient atrioventricular block is considered to be caused by subclinical myocarditis [28], while another case with transient complete cardiac block also developed transient S1Q3T3, suggesting that his arrhythmia may have been caused by transient pulmonary hypertension induced by acute respiratory distress syndrome [29].
3. Potential Mechanism of COVID-19 Complicated with Arrhythmia
Multiple trigger factors and potential mechanisms raise the risk of arrhythmias in patients with COVID-19. For COVID-19 patients with previous cardiovascular diseases, viral infection increases metabolic needs and makes chronic cardiovascular diseases unstable [30], thereby directly causing myocardial damage or inflammatory response and higher risk of arrhythmias or other heart injuries [31,32]. Current studies have suggested that patients with COVID-19 who do not have a history of cardiovascular disease also have arrhythmias and other heart damage [33,34].
In summary, current studies have shown that the occurrence of arrhythmias in patients with COVID-19 may be attributed to changes in angiotensin converting enzyme 2 expression, myocarditis, cytokine storm, hypoxemia, myocardial strain, electrolyte abnormalities, intravascular volume imbalance, drug interaction and stress response caused by viral infection (Figure 1).
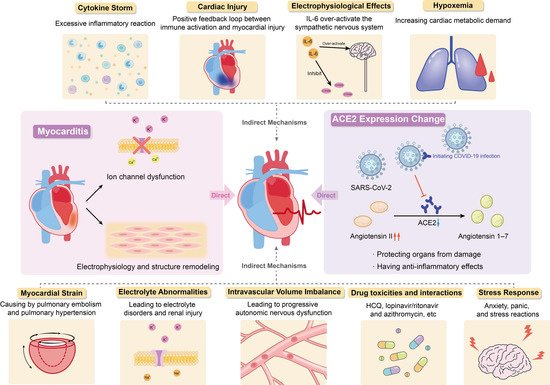
Figure 1. Potential Mechanism of COVID-19 Complicated with Arrhythmia.
3.1. Direct Mechanism
3.1.1. Changes in Angiotensin Converting Enzyme 2 Expression Induced by Viral Infection
The exact pathophysiological mechanism of SARS-CoV-2-induced arrhythmias is not completely clear, but the existing data show that SARS-CoV-2 may be able to invade the heart and directly induce cardiac injury and arrhythmias. This effect is mainly achieved through the interaction of angiotensin 2 (ACE2), ACE2 inhibitor/receptor blocker and COVID-19.
Angiotensin I is converted to angiotensin II through angiotensin enzyme (ACE), which can be inhibited by ACE inhibitors. Angiotensin I and angiotensin II can be converted into angiotensin 1–9 and angiotensin 1–7 by ACE2, respectively. They can antagonize the activation of renin-angiotensin-aldosterone system (RAAS), protect organs from injury, and have anti-inflammatory effects, especially in people with history of hypertension, diabetes, or cardiovascular disease. Angiotensin II plays an inflammatory role through the angiotensin type 1 (AT1) receptor, and the combination of the two can promote the degradation of ACE2. The AT1 receptor blockers (ARBs) can inhibit this binding to prevent ACE2 degradation. Long-term use of ARBs can convert angiotensin II to angiotensin 1–7, increase the expression of ACE2 and promote anti-inflammatory effects.
The angiotensin enzyme2 is highly expressed in lung (mainly type II alveolar cells) and heart tissues and is the receptor of SARS-CoV-2 invading host cells [12,35]. The SARS-CoV-2 replicates and proliferates in cells and downregulates the expression of ACE2, which has a direct effect on the myocardium. At the same time, the virus in cells also inhibits the formation of stress granules through its accessory proteins, promotes its replication and causes cell injury [36]. This may be one of the reasons for the adverse outcome of COVID-19 patients.
Previous cardiovascular diseases affect ACE2 levels through RAAS-related pathophysiology and RAAS inhibitors [37], which will not only increase the degree of COVID-19 entering the lung and heart but also have a direct impact on cardiovascular disease, leading to heart injury. At the same time, it is also related to the poor prognosis and higher risk of death in patients with COVID-19 [3,34]. High angiotensin II levels in the circulatory system can lead to inflammation, vasoconstriction, myocardial injury, and thrombosis [33,38,39].
Therefore, ACE2 has dual effects. On the one hand, due to the high expression of ACE2 in alveolar cells and can protect lung function, long-term use of RAAS inhibitors in patients with cardiovascular disease may lead to ACE2 activity increased, which is beneficial to patients with COVID-19. On the other hand, ACE2 can initiate SARS-CoV-2 infection and then lead to downregulation of ACE2 and the toxicity of excessive accumulation of angiotensin II, gradually resulting in ARDS and fulminant myocarditis. These two effects are antagonistic to each other. Inhibition of RAAS leads to the upregulation of ACE2, which makes patients susceptible to COVID-19 but at the same time weakens the downregulation of ACE2 caused by viral infection and reduces toxicity, thus reducing the cardiovascular complications of COVID-19 patients [37,40]. At present, the potential benefits and disadvantages of ACE inhibitors/ARBs are still controversial.
3.1.2. Myocarditis
Some COVID-19 patients die of fulminant myocarditis or viral activated cytokine storms, indicating that COVID-19 may cause a strong inflammatory response and increase myocarditis-related cardiac injury [41]. Viral infection is one of the leading causes of myocarditis and is also recognized in COVID-19. To date, there have been several relevant literature reports on SARS-CoV-2-induced myocarditis [42,43]. It is important to note that direct detection of the virus in the heart is only rarely reported, localization of SARS-CoV-2 is mostly confined to interstitial cells or macrophages, rather than the cardiomyocytes, meanwhile, data from Lindner et al. suggest that the presence of viruses in cardiac tissue does not necessarily cause myocarditis associated with inflammatory cell infiltration [44,45]. Recently published literature has found that the prevalence of myocarditis in autopsy cases is less than 2% [46], and the US Centers for Disease Control and Prevention has reported that although the risk of myocarditis is nearly 16 times higher in patients with COVID-19 than in those without COVID-19 and this risk is age-related, the prevalence of myocarditis in patients with COVID-19 is less than 0.2% of all cases, which is apparently very low [47]. The rate of cardiac injury in COVID-19 patients varied considerably between studies (5–40%), but overall there was a high prevalence of cardiac injury in COVID-19 patients, which is unlikely to be due to myocarditis that has a low prevalence [48]. Myocarditis associated with viral invasion, ion channel dysfunction, electrophysiology and structural remodeling caused by host and viral factors may lead to fatal arrhythmias. The mechanism is to induce abnormal calcium treatment and downregulation of potassium channel expression, resulting in prolonged repolarization time and abnormal conduction [49].
In addition, it has been reported that patients with higher levels of cardiac troponin (Tn) have a higher incidence and mortality of malignant tachyarrhythmia [19]. A study of 187 patients with COVID-19 found that 27.8% of the patients had myocardial damage, among which patients with potential cardiovascular disease with elevated troponin T (TnT) had the highest mortality, followed by patients without history of cardiovascular disease but with elevated TnT, and patients with normal TnT had the lowest mortality [19]. Serum cardiac troponin I (cTnI) levels are increased in hospitalized patients and can be used as an indicator of severe SARS-CoV-2 infection and a marker of complications caused by SARS-CoV-2 infection [50]. When the level of serum hypersensitive cardiac troponin I (hs-TnI) exceeds the upper limit of the reference range, cardiac injury is considered to occur (>28 pg/mL) [51]. A study of 671 cases showed that the level of high sensitivity plasma troponin I (hs-TnI) in 15.8% of COVID-19 patients was higher than that in the normal range, while the average level of hs-TnI in patients who died was significantly higher than that in survivors [52]. Patients with malignant tachyarrhythmia associated with elevated troponin levels should be highly suspected of having potential myocarditis [53]. The increase in troponin is often accompanied by an increase in inflammatory markers, which also suggests the role of cytokine storms in arrhythmias [54].
3.2. Indirect Mechanism
3.2.1. Cytokine Storm
Some researchers have suggested that inflammatory cytokines may mediate the development of arrhythmias in COVID-19 patients, and that arrhythmias are not a direct result of SARS-CoV-2 infection, but rather a generalized response to the systemic inflammatory response to severe viral disease [23,55,56]. Postmortem examination showed that the myocardium of patients with COVID-19 was infiltrated by interstitial monocytes, especially in the case of fulminant myocarditis [57,58]. It can be speculated that arrhythmia is related to cytokine storm, and its essence is excessive inflammatory reaction. However, autopsy cases have also reported that the presence of the virus was not associated with increased infiltration of monocytes into the myocardium [44].
The virus entered type II alveolar cells and replicated at the early stage of SARS-CoV-2 infection. The body activates the immune inflammatory response under the effect of viral cytotoxicity, which leads to ARDS and hypoxia. In addition, the level of inflammatory cytokines in the plasma of patients in ICU and arrhythmia incidence were higher than that of patients not in ICU [19,50,59], inflammatory cytokines, such as IL-6 and IL-8, are associated with lung injury and poor prognosis in patients with COVID-19, and IL-6 and IL-10 are significantly elevated in patients with severe COVID-19 pneumonia, suggesting that the inflammatory response was related to the severity of infection and impacts arrhythmias occurrence [55]. If the virus is not effectively cleared at this stage, an excessive inflammatory response will occur. At this time, the cytokine storm will lead to multiple organ dysfunction and then arrhythmia [60]. The report showed that Th17 cells increased, CD8+ T lymphocyte toxicity increased and migration to cardiomyocytes caused inflammation in patients with COVID-19 [57]. It can be inferred that in the stage of SARS-CoV-2 infection, immune system dysfunction leads to the release of inflammatory cytokines and severe acute systemic inflammation and cytokine storm responses, resulting in irreversible multiple organ damage, including cardiac dysfunction.
3.2.2. Cardiac Injury
It is well known that the inflammatory response helps to prevent infection, but when inflammatory cytokines are released excessively, an excessive inflammatory response will occur. Several studies have found that cardiac injury is common in patients with COVID-19 [61,62].
Heart damage may be a secondary manifestation of systemic infection. The release of inflammatory cytokines causes cytokine storm, which enhances the activation of T lymphocytes, thereby releasing more cytokines, forming a positive feedback loop between immune activation and myocardial injury [42]. This will strengthen myocardial injury and directly cause arrhythmias due to cellular lesions, as well as reduced coronary blood flow, reduced oxygen supply, unstable coronary plaques and microthrombosis [50]. This is also supported by the study by Xia et al. who found that elevated white blood cell counts, and elevated levels of inflammatory cytokines were more common in patients with cardiac injury and that inflammatory cytokine levels were positively associated with the risk of patients developing myocardial injury [62]. Meanwhile, the levels of circulating pro-inflammatory cytokines in COVID-19 patients are directly and independently correlated with troponin levels [63].
3.2.3. Electrophysiological Effects
Systemic inflammation rapidly induces cytokine-mediated electrical remodeling of the ventricles and significant QTc prolongation during acute infections [64]. Previous studies have found that IL-6 can directly block hERG channels in ventricular myocytes and over-activate the sympathetic nervous system [65,66]. A large cohort study showing an independent association between SARS-CoV-2 infection and QTc prolongation, IL-6 levels and QTc maximum in hospitalized COVID-19 patient were directly correlated, suggesting that IL-6 and QTc prolongation are related, but other factors may be involved [67]. Other studies have similar findings, with elevated IL-6 predicting the risk of arrhythmias in COVID-19 patients [55,68].
Tumor necrosis factor-α (IL-1), IL-6 (IL-6) and IL-1 (IL-1) have also been shown to regulate the function of ion channels and prolong the time of ventricular action potential by interacting with potassium and calcium channels [64,69,70]. Moreover, elevated cytokines (especially IL-6) also reversibly downregulate the expression of connexin43 in the atria and atrioventricular node, rapidly inducing cardiac electrical remodeling and increasing atrial fibrosis, thereby promoting the development of atrial fibrillation and AV block [71,72].
In addition, increased IL-6 during systemic inflammation can inhibit cytochrome P450, especially CYP3A4, which may lead to the QT prolongation and increase the danger of torsional ventricular tachycardia at the tip [73,74,75]. The SARS-CoV-2 had subtype-specific effects on CYP, such as reduced activity of CYP1A2, CYP2C19 and CYP3A and increased activity of CYP2B6 and CYP2C9 [75,76].
Besides effects on CYP, other electrophysiological effects caused by cytokines also have effects on the organism. The presence of fever in COVID-19 patients may promote the development of LQTS by altering K+ dependence [77]. The inflammatory response releases large amounts of inflammatory cytokines that act on the hypothalamus, leading to activation of the sympathetic nervous system via the central pathway and consequent arrhythmias [78]. Inflammatory factors acting on the peripheral pathway lead to increased left stellate ganglion remodeling, which also leads to the same result [79]. In the male, a systemic inflammatory response is associated with reduced testosterone levels and transient inflammatory hypotestosteronemia, which is significantly associated with an increased risk of long QT syndrome and TdP [80].
3.2.4. Hypoxemia
The SARS-CoV-2 infection can cause severe lung injury, resulting in acute respiratory failure and myocardial damage. According to a previous study, 32% of COVID-19 patients develop different degrees of hypoxemia [81]. Up to 76% of patients need oxygen support [59]. Hypoxemia may increase pulmonary artery pressure and right ventricular afterload, increase cardiac metabolic demand, affect myocardial oxygen supply, and generate myocardial injury [82,83]. The study by Guo T. et al. also confirmed that COVID-19 patients with myocardial injury have worse hypoxemia compared to those without myocardial injury [19].
3.2.5. Myocardial Strain
Pulmonary embolism is one of the most common thrombotic complications in COVID-19 patients [84], and patients presenting with ARDS can develop secondary pulmonary hypertension. Pulmonary embolism and pulmonary hypertension may cause myocardial strain. On the one hand, a COVID-19 patient with transient pulmonary hypertension developed transient complete cardiac block [29]. On the other hand, patients with pulmonary hypertension and hypoxemia have higher risk of atrial tachyarrhythmia on account of right atrial pressure and sympathetic nerve excitement [85]. The degree of myocardial strain is associated with the development of QTc prolongation in COVID-19 patients, which can be indicated by circulating BNP levels [86].
3.2.6. Electrolyte Abnormalities
Arrhythmia caused or aggravated by abnormal electrolytes in patients with COVID-19 has been reported to a certain extent [87,88]. Electrolyte disturbances are common in COVID-19 patients, particularly hypokalaemia, hyponatraemia and hypocalcaemia. In a case series containing 416 patients, 7.2% of patients were reported electrolyte disorders [89]. Altered function of the RAAS system, gastrointestinal dysfunction, inflammatory factors and secondary renal tubular dysfunction caused by viral invasion may all contribute to electrolyte imbalance [90]. A retrospective study of 85 patients showed that 27% of patients had acute renal injury [91].
This entry is adapted from the peer-reviewed paper 10.3390/jcdd9090292
This entry is offline, you can click here to edit this entry!