Cachexia is a metabolic syndrome consisting of massive loss of muscle mass and function that has a severe impact on the quality of life and survival of cancer patients. Up to 20% of lung cancer patients and up to 80% of pancreatic cancer patients are diagnosed with cachexia, leading to death in 20% of them. The main drivers of cachexia are cytokines such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), macrophage inhibitory cytokine 1 (MIC-1/GDF15) and transforming growth factor-beta (TGF-β). Besides its double-edged role as a tumor suppressor and activator, TGF-β causes muscle loss through myostatin-based signaling, involved in the reduction in protein synthesis and enhanced protein degradation.
- cachexia
- TGF-β
- cancer-related syndrome
1. Introduction
2. TGF-β Signaling Activation
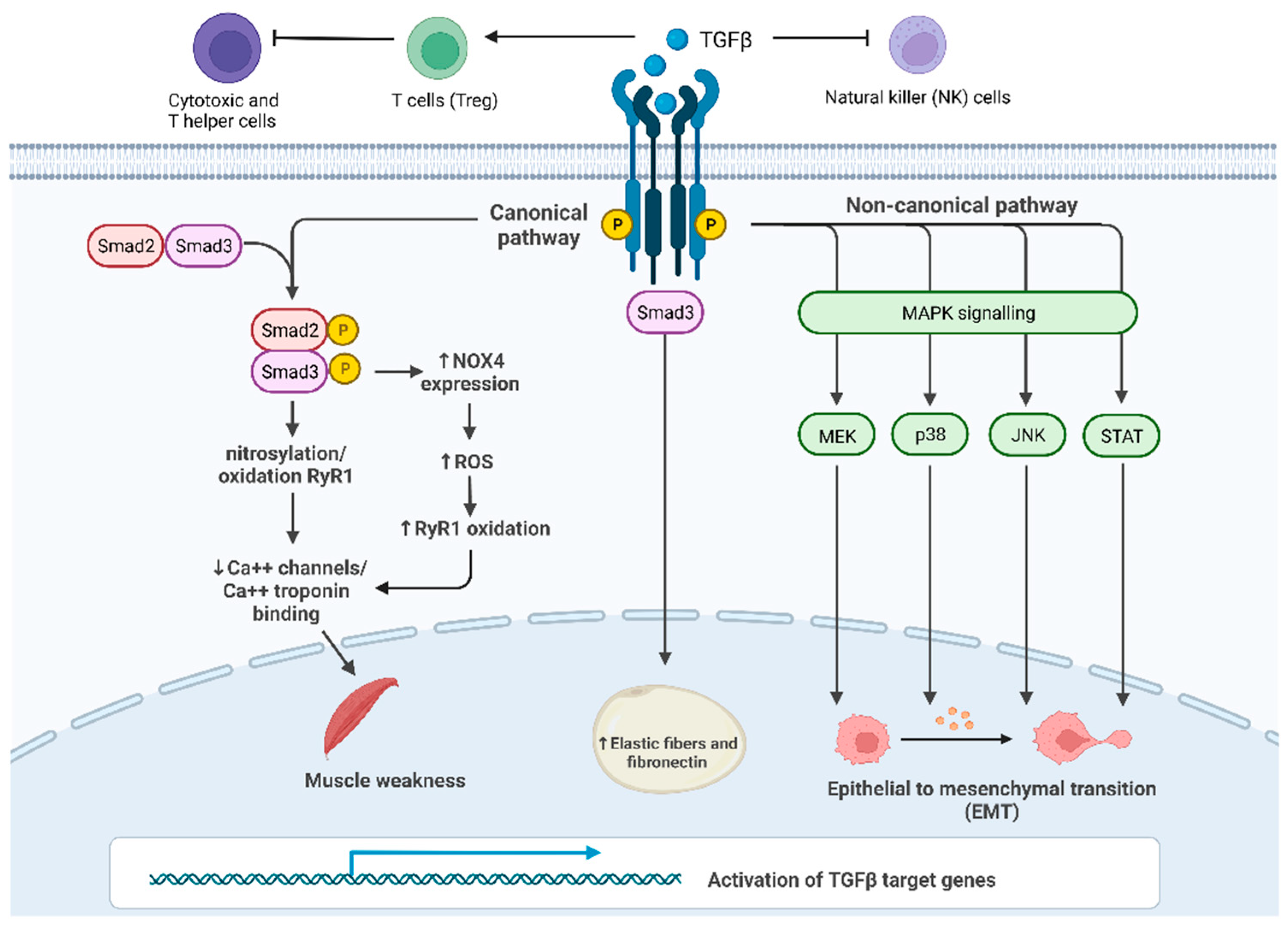
This entry is adapted from the peer-reviewed paper 10.3390/cells11172671
References
- Tisdale, M.J. Molecular Pathways Leading to Cancer Cachexia. Physiology 2005, 20, 340–348.
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and Classification of Cancer Cachexia: An International Consensus. Lancet Oncol. 2011, 12, 489–495.
- Tisdale, M.J. The “Cancer Cachectic Factor. ” Supportive Care Cancer 2003, 11, 73–78.
- Anker, M.S.; Holcomb, R.; Muscaritoli, M.; von Haehling, S.; Haverkamp, W.; Jatoi, A.; Morley, J.E.; Strasser, F.; Landmesser, U.; Coats, A.J.S.; et al. Orphan Disease Status of Cancer Cachexia in the USA and in the European Union: A Systematic Review. J. Cachexia Sarcopenia Muscle 2019, 10, 22–34.
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer Cachexia: Understanding the Molecular Basis. Nat. Rev. Cancer 2014, 14, 754–762.
- Prado, C.M.M.; Bekaii-Saab, T.; Doyle, L.A.; Shrestha, S.; Ghosh, S.; Baracos, V.E.; Sawyer, M.B. Skeletal Muscle Anabolism Is a Side Effect of Therapy with the MEK Inhibitor: Selumetinib in Patients with Cholangiocarcinoma. Br. J. Cancer 2012, 106, 1583–1586.
- Pin, F.; Couch, M.E.; Bonetto, A. Preservation of Muscle Mass as a Strategy to Reduce the Toxic Effects of Cancer Chemotherapy on Body Composition. Curr. Opin. Support Palliat Care 2018, 12, 420–426.
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-Associated Cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105.
- Lippitz, B.E.; Harris, R.A. Cytokine Patterns in Cancer Patients: A Review of the Correlation between Interleukin 6 and Prognosis. Oncoimmunology 2016, 5, e1093722.
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of Tumor Microenvironment in Tumorigenesis. J. Cancer 2017, 8, 761–773.
- Landskron, G.; de La Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 149185.
- Fearon, K.C.H.; Glass, D.J.; Guttridge, D.C. Cancer Cachexia: Mediators, Signaling, and Metabolic Pathways. Cell Metab. 2012, 16, 153–166.
- Tsai, V.W.W.; Lin, S.; Brown, D.A.; Salis, A.; Breit, S.N. Anorexia-Cachexia and Obesity Treatment May Be Two Sides of the Same Coin: Role of the TGF-b Superfamily Cytokine MIC-1/GDF15. Int. J. Obes 2016, 40, 193–197.
- Chen, J.L.; Walton, K.L.; Hagg, A.; Colgan, T.D.; Johnson, K.; Qian, H.; Gregorevic, P.; Harrison, C.A. Specific Targeting of TGF-β Family Ligands Demonstrates Distinct Roles in the Regulation of Muscle Mass in Health and Disease. Proc. Natl. Acad. Sci. USA 2017, 114, E5266–E5275.
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of Skeletal Muscle Mass in Mice by a New TGF-Beta Superfamily Member. Nature 1997, 387, 83–90.
- Mendias, C.L.; Gumucio, J.P.; Davis, M.E.; Bromley, C.W.; Davis, C.S.; Brooks, S.V. Transforming Growth Factor-Beta Induces Skeletal Muscle Atrophy and Fibrosis through the Induction of Atrogin-1 and Scleraxis. Muscle Nerve 2012, 45, 55–59.
- Greco, S.H.; Tomkötter, L.; Vahle, A.K.; Rokosh, R.; Avanzi, A.; Mahmood, S.K.; Deutsch, M.; Alothman, S.; Alqunaibit, D.; Ochi, A.; et al. TGF-β Blockade Reduces Mortality and Metabolic Changes in a Validated Murine Model of Pancreatic Cancer Cachexia. PLoS ONE 2015, 10, e0132786.
- Farkas, J.; von Haehling, S.; Kalantar-Zadeh, K.; Morley, J.E.; Anker, S.D.; Lainscak, M. Cachexia as a Major Public Health Problem: Frequent, Costly, and Deadly. J. Cachexia Sarcopenia Muscle 2013, 4, 173–178.
- Roeland, E.J.; Bohlke, K.; Baracos, V.E.; Bruera, E.; Fabbro, E.D.; Dixon, S.; Fallon, M.; Herrstedt, J.; Lau, H.; Platek, M.; et al. Management of Cancer Cachexia: ASCO Guideline. J. Clin. Oncol. 2020, 38, 2438–2453.
- Chen, W.J.; Wahl, S.M. TGF-Beta: The Missing Link in CD4+CD25+ Regulatory T Cell-Mediated Immunosuppression. Cytokine Growth Factor Rev. 2003, 14, 85–89.
- Colak, S.; ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71.
- Tian, M.; Neil, J.R.; Schiemann, W.P. Transforming Growth Factor-β and the Hallmarks of Cancer. Cell Signal. 2011, 23, 951–962.
- Dardare, J.; Witz, A.; Merlin, J.L.; Gilson, P.; Harlé, A. SMAD4 and the TGFβ Pathway in Patients with Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 3534.
- Qiu, T.; Wu, X.; Zhang, F.; Clemens, T.L.; Wan, M.; Cao, X. TGF-Beta Type II Receptor Phosphorylates PTH Receptor to Integrate Bone Remodelling Signalling. Nat. Cell Biol. 2010, 12, 224–234.