Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Plant Sciences
Cannabis (Cannabis sativa L.), also known as hemp, is one of the oldest cultivated crops, grown for both its use in textile and cordage production, and its unique chemical properties.
- cannabis
- genomics
- metabolomics
- multi-omics
1. Introduction
Cannabis (Cannabis sativa L.) is an herbaceous species originating from Central Asia and is distributed to lesser extent all over the world, growing in wide ranging habitats and climatic conditions [1].
Cannabis is considered one of the oldest cultivated multipurpose crops. In fact, it can be classified as fibre crop (hemp), with a long history of rope and textile making, thanks to its cellulosic and woody fibers, and also a drug crop (medicinal cannabis) since it is used for therapeutic purposes [2]. Hemp contains less than 0.3% of tetrahydrocannabinol (THC), whereas medicinal cannabis contains a greater amount of this metabolite, generally accounting for up to 5% of the dry weight [3]. THC, along with cannabidiol (CBD), are the most important secondary metabolites produced by cannabis and, among the ~130 secondary metabolites identified [4,5], they are the predominant focus of breeding programs and pharmaceutical industries [2,6]. Abbreviations used throughout the manuscript are listed in Table 1.
Table 1. List of abbreviations used in this research.
| Abbreviations | Definition |
|---|---|
| CBC | Cannabichromene |
| CBCA | Cannabichromenic Acid |
| CBD | Cannabidiol |
| CBDA | Acidic Precursors of Cannabidiol |
| CBG | Cannabigerol |
| CBGA | Acidic Precursors of Cannabigerolic |
| CBDRx | High Cannabidiol Content Cannabis Cultivar |
| CNV | Copy Number Variation |
| DEG | Differentially Expressed Gene |
| DLLLME | Dispersive Liquid-Liquid Microextraction |
| DMAPP | Dimethylallyl Diphosphate |
| FN | Finola Cannabis Cultivar |
| GBS | Genotype By Sequencing |
| GC-EIMS | Gas Chromatography-Electron Impact Mass Spectrometry |
| GC-FID | Gas Chromatography-Flame Ionization Detection |
| GC-MS | Gas Chromatography–Mass Spectrometry |
| GPP | Geranyl Diphosphate |
| GS | Genomic Selection |
| GWAS | Genome-Wide Association Study |
| HPLC | High Performance Liquid Chromatography |
| HRMS | High-Resolution Mass Spectrometry |
| HS-SPME | Headspace Solid-Phase Micro Extraction |
| IPP | Isopentenyl Diphosphate |
| ISO-Seq | Isoform Sequencing |
| JL | Jamaican Lion Cannabis Cultivar |
| LC-DAD | Liquid Chromatography-Diode Array Detector |
| LC-MS | Liquid Chromatography–Mass Spectrometry |
| MEP | Methylerythritol Phosphate |
| MS | Mass Spectrometry |
| NGS | Next Generation Sequencing |
| NMR | Nuclear Magnetic Resonance |
| OA | Olivetolic Acid |
| ORF | Open Reading Frames |
| PAVs | Presence/Absence Variations |
| PPFD | Photosynthetic Photon Flux Density |
| PK | Purple Kush Cannabis Cultivar |
| QTL | Quantitative Trait Loci |
| QQQ | Triple-Quadrupole Mass Spectrometry |
| qRT-PCR | Real time/Quantitative PCR—Polymerase Chain Reaction |
| RNA-Seq | RNA-Sequencing |
| ROS | Reactive Oxygen Species |
| SFE | Supercritical Fluid Extraction |
| SMRT | Single-Molecule Real-Time Sequencing |
| SNPs | Single Nucleotide Polymorphisms |
| SPE | Solid Phase Extraction |
| SPME | Solid Phase Micro Extraction |
| SVs | Structural Variants |
| THC | Tetrahydrocannabinol |
| THCA | Acidic Precursors of Tetrahydrocannabinol |
| WGCNA | Weighted Gene Co-expression Network Analysis |
| WGS | Whole Genome Shotgun Sequencing |
Despite their similar chemical structures, these two metabolites do not have the same effects on the human body [2]. THC is psychoactive and induces a sense of euphoria, while CBD is not, but instead has therapeutic uses in reducing anxiety and depression [2].
In spite of the small size of the genus, the exact number of cannabis species is still not well defined. According to scientific studies, there are three cannabis species with distinct phenotypic differences, namely C. sativa L., C. indica Lam (Lamarck), and C. ruderalis [7,8]. However, the majority of classifications performed to date evidence the existence of C. sativa and C. indica only. Specific crosses between these two species are referred to as hybrids and have highly variable phenotypes, showing intermediate features to the parents [9]. However, these common distinctions are not representative of the evolutionary relationships [10]. There is a relevant phenotypic variation among these cannabis species, especially involving cannabinoids [11,12] and terpenoids levels [13,14], as well as differences among genotypes [7,9,15], and morphologic features like flowering time, and branch and internode length [1]. Morphologically, C. sativa plants are tall, have less dense buds, narrow leaves, and produce high levels of THC. Conversely, C. indica Lam plants are short with denser buds and broader leaves, and they synthetise high levels of both THC and CBD [1,9,11].
Within a given cannabis species, cultivars are categorised into groups based on their chemotype, from I to V, according to the number and ratio of main cannabinoids [16]. These compound profiles can be employed both as quality markers and fingerprints for cannabis standardization.
Cannabis growth can be divided into four distinct phases: germination, seedling, vegetative and flowering stage. Each phase is characterised by its own photoperiod, environmental and nutritional needs [17]. Cannabis germinates, reaches maturity, reproduces and dies in one year in the wild. The flowers are unisexual, and therefore male and female individuals are distinct. However, hermaphrodites have often been documented [17,18]. In general, males and females are not identified until the second week of the bloom cycle. Since only female inflorescences are used to produce extracts, once a male plant is identified, it is generally discarded. The vegetative phase is characterised by the greatest increase in biomass and overall growth. During this phase, roots extend considerably, leaves start growing and expanding to increase the photosynthetic area, and transpiration rises, so water intake increases as well. The reproductive phase of cannabis development involves massive hormonal changes induced by photoperiod and this can be enhanced by an increase in red and far-red wavelengths of light [17,18]. In this phase, the first major increase in cannabinoid levels occurs in female inflorescences [17,18].
Due to the legislation regulating cannabis cultivation, the cannabinoid biosynthetic pathway has not been characterized in detail, especially from a molecular and genetic perspective [19]. Conversely, many other major crop species have already been widely studied, especially after the development of next generation sequencing (NGS) technologies [19,20]. The recent relaxation in legislation [21,22], as well as the availability of the cannabis genomic sequence, consisting of a complex genome, containing 843 Mb and 818 Mb for male and female plants, respectively [23], is now facilitating research on this crop. The diploid genome consisting of nine autosomes and a pair of sex chromosomes [24,25], is highly heterozygous, and contains many repetitive elements (~70%) [5,26,27]. Despite the presence of distinct sex chromosomes, some external factors, such as a shorter photoperiod, a lower temperature, and the application of chemicals, such as ethylene inhibitors, on leaves, can enhance pollen production in female flowers, resulting in ‘feminised seeds’ [28]. This technique has been often used in cannabis breeding to generate target populations to investigate key phytochemical and qualitative traits.
2. Studying the Metabolomic Profile of Cannabis
2.1. Key Metabolites: An Overview
Cannabis is a polymorphic plant species producing a diverse profile of bioactive metabolites which have unique chemical structures and physiochemical properties [20]. Among them, the main compound class are the cannabinoids, accounting for ~20% of the total secondary metabolites in cannabis, and terpenoids are also highly abundant, of which isoprenes, monoterpenes, and sesquiterpenes are predominant [32].
Cannabinoids are primarily synthesized in the glandular trichomes of female flowers, while trichomes of male flowers are generally very low in cannabinoids [33]. Cannabis trichomes are classified as stalked, sessile, or bulbous, where bulbous trichomes produce limited cannabinoids compared to the other types [34]. Trichomes contain resin storage cells and, during the flower and seed maturation stage, the composition of cannabinoids within the resin changes, reaching the highest level at flower maturity [33]. The concentration of cannabinoids increases in warmer temperatures but is negatively correlated with the mineral content of soil [35]. Cannabinoid yield is also affected by UV-radiation and an increase was observed in cannabis flowers after UV-B-induced stress [36].
Figure 1 illustrates the main steps of THC and CBD biosynthesis. THC and CBD are synthesised from two distinct metabolic pathways: the polyketide and the methylerythritol phosphate (MEP), producing olivetolic acid (OA) and geranyl diphosphate (GPP), respectively [5,37]. Specifically, OA and GPP synthesize the cannabigerolic acid (CBGA), containing a pentyl side chain, which produces the acidic precursors of THC (THCA) and CBD (CBDA) [38]. The cannabichromenic acid (CBCA) is also produced [39]. Synthesis of THCA, CBDA, and CBCA proceeds through the appropriate oxidocyclases: THCA synthase, CBDA synthase, and CBCA synthase, respectively.
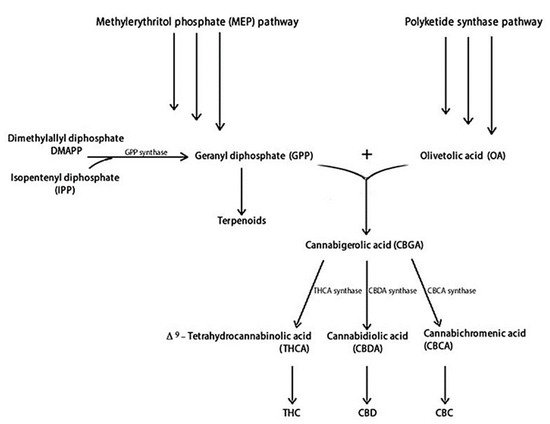
These acidic cannabinoids are thermally unstable and can be decarboxylated when exposed to light or heat via smoking [40]. Terpenoids are produced by dimethylallyl diphosphate (DMAPP) and isopentenyl diphosphate (IPP) metabolic pathways, which share a GPP precursor with cannabinoids [41].
However, the biosynthesis of cannabinoids and terpenes is still far from being fully understood at the molecular level [34]. Further and innovative investigations are crucial for many upcoming medicinal cannabis applications, where novel bioactive compounds or less abundant cannabinoids may be of great interest [42].
2.2. Cannabis Metabolite Profiling Techniques
The chemical composition of cannabis is extremely important. In fact, it can be unique for each cultivar and the metabolic fingerprint is fundamental to exploring the differences among them [43].
A variety of techniques have been employed to extract and analyse compounds from cannabis, and different methods are used depending on whether the aim is to investigate cannabinoids or terpenes [44].
The most popular platforms used to analyse cannabinoids are gas chromatography (GC) and liquid chromatography (LC), coupled with mass spectrometry (MS) [44,45]. LC is used for the analysis of non-volatile and thermally labile compounds, while GC allows the analysis of thermally stable molecules and often derivatization agents are used to aid this process. GC-MS usually uses electron ionization (EI) to fragment the analytes in a consistent way, whereas LC-MS generates ions with less diagnostic fragmentation information. Considering the complex metabolomic matrix of cannabinoids, working on less abundant, more novel cannabinoids is challenging [44]. Cannabis testing laboratories often prefer to use LC for cannabinoid chemical analysis, due to simpler sample preparation steps. For instance, the derivatization and decarboxylation of related precursory molecules, which are necessary for GC based methods, can be skipped with LC [44].
GC-MS was employed in an interesting study recently [46] where the effects of natural and artificial lighting on cannabinoid metabolism were analyzed. Specifically, treatments of cannabis crops with 3 different light spectra, high-pressure sodium (HPS), AP673L (LED), and NS1 (LED) were investigated [46]. Results explored how these treatments affected cannabis morphology and its CBG, CBD and THC content, but they only had a minor impact on the overall yield. Furthermore, LED lights resulted in higher amounts of plant growth and improved the cannabinoid profile, when compared to HPS lights. Plants grown under LED light conditions had a boosted THC and CBD concentrations, and cannabis cultivars having an elevated THC yield also exhibited a higher photosynthetic capacity. This suggests that different cannabis chemovars may be optimally cultivated under different light intensities. The potential of LED lighting in the cannabis sector has been further investigated, but there is a lack of tangible evidence on how light quality and light source affects extract quality and yield [36]. Furthermore, a recent study based on gas chromatography electron impact mass spectrometry (GC-EI-MS) investigated the lipids extracted from seeds of C. sativa and identified over 40 cannabinoids. Indeed, 16 of which had never been detected before, and some were hoped to have future medicinal potential [47].
High Performance Liquid Chromatography (HPLC) has also been widely applied in the study of cannabis metabolites, with it being faster, more sensitive and efficient compared to LC. It was used in a study in which a set of cannabis varieties, representative of all chemotypes, were analysed and compared [48]. The total yield of the major cannabinoids CBD, CBG and THC were measured in female monoecious hemp inflorescences. The varieties with the highest CBD content were ‘CS’ and ‘Carmagnola’, while the lowest amount of CBD was found in ‘Santhica 27’. Conversely, ‘Bernabeo’ genotype showed the highest value of CBG and, as expected, the THC content of the medical varieties, like ‘CINBOL’ were very high. Another study based on HPLC showed that the content of cannabinoids is highly influenced by the cultivar and the plant growth stage [49]. Specifically, the investigation was focused on a set of industrial hemp cultivars, and the results demonstrated that, although some of them, e.g., ‘Futura75’, ‘Fédora17’, ‘Félina32’, and ‘Ferimon’, are mainly cultivated for fibre and seed production, they can also be used for cannabinoids extraction. Furthermore, these cultivars showed a maximum CBDA yield when the seed completed its maturation.
Besides cannabinoids, terpenes are another important compound class in cannabis. They are usually analysed by GC, coupled with various detectors, such as Flame Ionization Detection (FID), which is the most commonly applied due to its low cost and ease of use. A study based on this platform allowed the classification of 13 cannabis cultivars based on their terpenoid profile [50]. Specifically, results highlighted how some cultivars fit into one or more specific chemotypes, whereas in other cultivars this association is not so clear-cut [50]. More recently, by taking a combined analysis approach using both a LC-diode array detector (LC-DAD) and GC-FID technologies, further classifications based on cannabinoid and terpenoid contents were proposed [43]. The results confirmed how the chemical composition is specific for each cultivar.
Depending on analytical aims, other extraction protocols can be applied to analyse cannabis metabolites, including supercritical fluid extraction (SFE), an extraction method using supercritical CO2 which is less expensive and more effective compared to chemical solvents, and represents a valid alternative to classic extraction systems [51]. For instance, sequential SFE and solid phase extraction (SPE) processes allowed THC to be extracted at a purity level suitable for quality control, where SPE was used as a purification technique for THC [52].
Furthermore, triple-quadrupole mass spectrometry techniques, known as QQQ and based on tandem MS, in which the first and third quadrupoles act as mass filters and the second fragments the chemical component, have also been applied to cannabis [53]. Due to its improved selectivity and sensitivity, this method was proven to be highly effective in quantitative cannabis metabolite analyses [53]. Therefore, it would also be possible to use this method to evaluate the abundance of cannabinoids in different parts of the crop, which may not be detected with other approaches given their low abundance [54].
Another versatile method increasingly used for the detection of analytes in complex matrices is the nuclear magnetic resonance spectroscopy (NMR) [55]. This technique is characterized by a low sensitivity compared to MS, even if it provides more reliable metabolite structure and does not require destructive sample preparation [56]. Furthermore, NMR allows simultaneous identification of multiple analytes. Despite these potential advantages, NMR has only been rarely applied in the detection of cannabinoids: as far as we know, it was mainly used to the authentication of hemp varieties [57,58].
In cannabis, due to the complex metabolome, the combination of several analytical methods usually gives the most comprehensive picture [2]. For instance, LC/QQQ/MS and NMR metabolomics analyses revealed the presence of several cannabinoids detected in extracts of cells of capitate-sessile and capitate-stalked trichomes as well [59]. Extracting and analysing the chemical profile of specific trichome lines holds great potential for use in future multi-omics experiments, as transcriptomic analyses could also be performed on these specific cell types [33]. The relationship between compound profile within these cells and their relative gene expression of cannabinoid biosynthetic genes could yield exciting new insights.
Another powerful technique used for the analysis of metabolites in cannabis is High-resolution mass spectrometry (HRMS), this technique enables a more precise identification of compounds with the same nominal mass due to the improvement in the calculated mass to charge ratio (m/z) to several decimal places compared to conventional MS. The use of this technique was found to have a great potential in the definition of cannabis chemovars [60]. Indeed, a recent study employed this method [61]: by using data from 20 varieties of C. sativa and a combined LC-HRMS platform, metabolites were mapped and annotated, and cannabis characteristic markers identified. The results of this approach were compared with those based only on major cannabinoid quantification, and it was found that minor compounds were highly predictive markers for differentiating cannabis varieties. Furthermore, these findings may be combined with other data coming from multi-level omics investigations, confirming the applicability and the potential of metabolomics in the understanding of cannabis metabolism regulation mechanisms.
3. Genomic and Transcriptomic Cannabis Profiles
Initial attempts to assemble the complex cannabis genome relied on the use of short-read sequencing technologies, but only recently have third-generation long-read sequencing technologies, such as Single-Molecule Real-Time (SMRT) sequencing (PacBio) and Oxford Nanopore Technologies (MinION) [25], led to an improved contiguity of reference sequences and correctly assembled ambiguous, highly repeated regions [25,62].
These advances resulted in the creation of four assemblies related to different cultivars: ‘Purple Kush’ (‘PK’, a drug type Cannabis), ‘Finola’ (‘FN’; a fiber type Cannabis), ‘Jamaican Lion’ (‘JL’; a wild accession) and ‘CBDRx’ (‘cs10’; with high CBD content) [27,63]. Comparisons of the transcriptome of ‘PK’ with that of the hemp cultivar ‘FN’ revealed that many genes encoding proteins involved in cannabinoid pathways are more highly expressed in ‘PK’ than in ‘FN’. Subsequently, these reference assemblies were annotated with full-length male and female mRNA sequencing to provide better information about isoforms complexity, genes and Y chromosome identification [25]. To date, the ‘cs10’ genome sequence assembly is considered to be the most complete and contiguous genome and is broadly used as the reference genome for cannabis [64].
The presence of copy number variations (CNVs) in cannabinoid synthases have been demonstrated in several cannabis genome studies [25,63] while the relation between cannabinoid synthase CNVs and cannabinoid content is still not clear [19]. Furthermore, highly similar loci are not adequately differentiated in short read sequencing approaches [19,65].
The availability of a sufficiently complete genome is crucial for the understanding of the cannabinoid pathways through a better knowledge of the underlying genes. Cannabinoid biosynthesis was investigated at molecular level, and several genes involved in this pathway were identified [48]. Furthermore, it was found that each gene consists of a single exon, with THCA synthase (THCAS) and CBCA synthase (CBCAS) sharing over 90% homology at amino acid level and over 80% homology to CBDAS [62,66].
The availability of these more complete genomic resources allowed the identification of sex chromosomes and of approximately 3500 gender-specific genes in the cannabis genome [25,67]. THCA and CBDA were found to be mainly produced in the inflorescences of female cannabis crops [24,25]. Thus, detecting male and female plants at early growth stages can increase yield and help design more specific cannabis breeding programs.
The association between THCA and CBDA synthase sequences and Quantitative Trait Loci (QTL) has been also reported [8,38]. An early study, using bi-parental mapping populations coming from a cross between hemp and drug cannabis, identified QTL regulating biochemical traits. Results suggested that THCA and CBDA synthase sequences are associated to a single multiple linked QTL [38]. Another study used a set of over 20 highly informative single nucleotide polymorphisms (SNP) markers related to cannabinoid and terpenoid expression to assess phylogenetic relationship, population genetics, and correlation with cannabis metabolites, demonstrating the utility of this method for efficient genotyping activities [8].
Other quite recent studies have been carried out, based on Genome Wide Association Studies (GWAS) in order to investigate novel cannabis genetic variants responsible for cannabis complex traits. For instance, candidate cannabinoid pathway genes have been identified, focusing on the alkyl side chain group whose genetic basis are mostly unknown although is a critical feature behind health properties [68]. These findings confirmed a previously known locus involved in cannabinoid synthesis pathway and other loci associated to chemotype variability [62], revealing 22 variants in a β-keto acyl carrier protein (ACP) reductase (BKR). It is worth noticing that genetic improvement of the alkyl side chain could help the development of new chemical chemotypes for pharmaceutical use. Furthermore, a GWAS approach has been applied to study the genetic architecture of flowering time and sex determination in hemp by using a panel of over 100 hemp accessions and a large set of SNP markers [69]. Several key genes and transcription factors involved in regulating phytohormones levels, like gibberellic acid, were identified in sex determination loci. These QTLs were proved to be responsible for the development of male flowers in female plants, being behind sex determination in monecious plants and its stability over time [69].
However, despite the advancement of the latest cannabis resources, the understanding of the genetic variation underlying complex agronomic traits of interest is still limited [19]. Although other recent investigations have been carried out, the development of efficient NGS tools and the construction of high-density genetic cannabis maps are necessary to improve the QTL mapping quality [62,63].
Genomic selection (GS) methods, which rely on genome-wide marker information to forecast the breeding impact of genotypes, could be a relevant approach to reach this aim. GS has been recently used in breeding other crop species, including rice and canola [70,71]. In both investigations, a multi-omics approach was applied to enhance agronomically important breeding traits including yield, grain weight and hybrid performance, underlining the advantages of combining omics datasets for GS analysis. In rice, the genomics predictions using genomic and metabolomic datasets showed better results than single omics approaches [70]. In canola, both SNPs and transcripts resulted reliable to predict hybrid performance using the most effective genomic unbiased prediction models. Compared to models just relying on pure genetic markers, those taking into account transcriptome data seem to be related to a significantly higher prediction accuracy, suggesting that transcripts contain relevant information beyond just genomic data [71]. The overall results reached to date are promising and open new perspectives for the genetic enhancement of complex traits regulated by a large number of genes. In the future, when further information become available and statistical models and phenotyping accuracy improve, these findings could also be applied to cannabis traits.
Regarding terpene synthase genes, the CsTPS family in the ‘JL’ reference genomes was characterised [72]. Copy number gains in CsTPS17 were observed in several cannabis cultivars, and CsTPS17 was identified as potentially involved in myrcene or limonene synthase [25]. In the same study, a copy number analysis in the ‘JL’ genome revealed a unique amplification of Gibberellic Acid Insensitive genes (GAI), which are known to be involved in plant growth. However, further investigations are required to better understand the contribution to yield of these genes.
Pathogen response genes, like those belonging to the Mildew Locus O (MLO) family, correlated with resistance to powdery mildew (PM), and the Thaumatin-Like Protein (TPLs) family, correlated with a wide range of pathogen resistance traits in plants including cannabis, were studied [25]. Specifically, the analysis confirmed extensive CNVs in cannabinoid synthesis and over 80 genes associated with resistance to Golovinomyces chicoracearum. Results also showed that plants with low THCA concentrations have a lower resistance to this pathogen. The antifungal response activity (against Fusarium oxysposum) of CsTLP1 was confirmed as well [25].
Gene expression investigations have been crucial for a better understanding of cannabinoid metabolic pathways [73]. A comparison of the transcriptomes of drug and fiber cannabis revealed that expression of the genes involved in the cannabinoid pathway is enhanced in drug cannabis [23], confirming that positive transcriptional regulators of the cannabinoid biosynthetic genes are more active in this variety. Furthermore, it was found that cannabis has more than 1220 transcription factors classified into families, such as MYB, bHLH, and AP2/ERF, which is considerable, but still far less than Arabidopsis, rice and maize [74]. A gland-specific transcription factor, HlWRKY1, controlling prenylated flavonoid and bitter acid biosynthesis in Humulus lupulus, a species closely related to cannabis, was detected [75]. Only recently THCA synthase promoters controlling expression exclusively in the trichomes were more thoroughly investigated [76]. Specifically, the CsAP2L1 (AP2-LIKE) and CsMYB1 (MYB) transcription factors were identified and the existence of a CsWRKY1 (WRKY) was confirmed. Results suggest that CsAP2L1 is a transcriptional activator, while CsMYB1 and CsWRKY1 are repressors. However, the understanding of transcriptional regulators that control THCA synthase expression and other cannabis metabolic pathways is still limited.
Further transcriptomic experiments were carried out on both fiber and medicinal cannabis crops [24,77]. Fiber cultivars were studied at different developmental stages, using samples from several stem regions, coming from the top, middle and bottom internodes of hemp stems [77]. Cell wall changes were correlated to RNA-Seq data and results showed that the major changes in fibers and gene expression occurred at the internodal regions and that each region of the stem presents a different gene expression profile. The gene ontology enrichment analysis underlined that genes related to the top region belonged to the DNA replication and cell cycle ontologies, the middle region was characterized by processes related to secondary cell wall biogenesis, while the bottom region was dominated by genes involved into phytohormone, as well as in secondary metabolic processes. Furthermore, immature stem tissue was characterized by photosynthesis related genes, along with others involved in the biosynthesis of specific secondary metabolites, mainly indole-containing compounds and oligolignols. Conversely, older, more mature internodes showed higher transcription levels of genes related to phytohormone production, as well as those involved in the lignification process.
In medicinal cannabis, genetic expression analysis in trichomes and leaf tissues facilitated the identification of many enzymes involved in the metabolic pathway of THCA and CBDA. In particular, a comprehensive transcriptome study using cannabis root, shoot, and flower was carried out [24]. There, genes involved in terpene and cannabinoid synthesis were detected and found to have high expression levels in trichomes. The results of this in-depth transcriptomic study represent a significant resource for future cannabis research. However, sparse information is still available today about the expression of genes associated with the synthesis of less abundant cannabinoids [47].
In the last few years, high-quality reference transcriptomes of two cultivars of Cannabis, a high THC cultivar and a CBD plus THC cultivar were assembled [65]. Each transcriptome contained over 20,000 protein-encoding transcripts. Transcripts for the cannabinoid pathway and related enzymes showed full-length open reading frames (ORFs) that align with the genomes of the ‘PK’ and ‘FN’ cultivars. Furthermore, two transcripts for OA were found to map to distinct locations on the ‘PK’ genome, suggesting that genes involved in OA biosynthesis are expressed in several cultivars.
Taking into account recent advances, transcriptomic studies have the potential to address one of the most crucial agricultural issues in many countries: the soil salinity, whose effects affect more than 800 million hectares worldwide [78]. Targeting breeding techniques to improve cannabis’s tolerance of saline and sodic soils is important to ensure high yields and maintain quality traits. A transcriptome of a saline-alkaline resistant cannabis, grown under NaHCO3 stress was investigated [79]. An RNA-Seq approach and weighted gene co-expression network analysis (WGCNA) were used to investigate the gene expression profiles and the results showed that some pathways, related to phenylpropanoid and sucrose, nitrogen, and amino acids biosynthesis, may be correlated to the response of cannabis under NaHCO3 stress. In the same year, key cannabis salt stress response genes were investigated by comparative transcriptome analyses of contrasting cannabis varieties, namely the W20 and K94 cultivars [80]. Over 80 differentially expressed genes (DEGs) which overlapped in the two cannabis varieties were identified, with more of these being up regulated than down regulated. Furthermore, results underlined how salt stress can induce increases in lipid peroxidation and reactive oxygen species (ROS) in cannabis and upregulate the expression of antioxidant genes as a response to ionic toxicity, as happens in other plants [81]. These DEGs represent potential targets for modern breeding techniques to adapt cannabis to grow efficiently on sodic soils, whilst still maintaining high chemical quality.
This entry is adapted from the peer-reviewed paper 10.3390/plants11162182
This entry is offline, you can click here to edit this entry!