Uveal melanoma (UM) is a poor prognosis cancer with no cure and limited treatment options [
1,
2,
3]. Originating in the eye, UM is the most common form of adult ocular cancer [
4]. Metastatic UM (MUM) is difficult to treat. A recent meta-analysis estimated a 60% relative survival rate of 15 to 20 years for patients after initial UM diagnosis [
5]. Approximately 50% of UM patients progress to MUM, with only 15% of patients reported to survive beyond one-year post diagnosis [
1,
4,
6]. In a meta-analysis (1 January 1980 to 29 March 2017) from a combined 2494 MUM patients, the median overall survival probability was 1.07 years across all forms of treatment [
7]. Common treatment strategies for MUM include chemotherapeutic agents, immunotherapy, site-directed (e.g., chemoembolization, immunoembolization) therapy and surgical tumor resection [
1,
2,
8,
9]. Additional targeted and combinatorial pharmacological-based treatment approaches such as receptor tyrosine kinases, MAPK pathway inhibitors, checkpoint inhibitors and histone deacetylase inhibitors have entered clinical trials [
9,
10,
11,
12].
A milestone in UM was achieved in January 2022 with FDA approval of Tebentafusp-tebn (KIMMTRAK
®), an immunotherapy, for treatment of HLA-A*02:01-positive adult patients with unresectable or metastatic UM [
13]. Very recently, Tebentafusp-tebn received EMA approval for use in the EU [
14,
15]. A phase III clinical trial (ClinicalTrials.gov Identifier: NCT03070392/EudraCT registration number: 2015-003153-18) reported that MUM patients (252) treated with Tebentafusp had improved one-year overall survival (OS) and six-months progression-free survival (PFS; 73% and 31%, respectively), on average, compared to patients (126) treated with control treatment (59% and 19%, respectively) options [
13,
16]. Overall it was reported that the patient cohort treated with Tebentafusp achieved 21.7 months median OS compared to 16 months (hazard ratio (HR) for death = 0.51; 95% CI 0.37 to 0.71;
p < 0.001) in the control cohort; median PFS was 3.3 months in Tebentafusp treatment group compared to 2.9 months (HR for disease progression or death = 0.73; 95% CI, 0.58 to 0.94;
p = 0.01) in the control group [
13]. However, at present this drug is only approved for use in a sub-cohort of patients, making it essential that more therapies and therapeutic targets are discovered for the treatment of a wider cohort of patients. Additionally, in an article published by Olivier and Prasad (2022), the authors put forth three key concerns regarding this study; (1) limited treatment options offered to control patient cohort in trial, (2) only 43% of the trial patients received treatment for disease progression post trial, and (3) interestingly, the observed median OS is
“far larger than, and disproportionate to the PFS benefit in terms of hazard ratios” and that mathematically, Tebentafusp
“is an outlier across trials in melanoma”, suggesting that further investigation is required for Tebentafusp [
17].
3. Selective HDAC6i as a Therapeutic Option for Uveal Melanoma
Interestingly, there are presently no registered clinical trials involving selective HDAC6i in UM/MUM, even though there are clinical trials ongoing for relapsed or refractory multiple myeloma, lymphoma, non-small cell lung cancer, metastatic breast cancer and solid tumor (
Table 1) [
23,
32,
33,
34]. Likewise, to date, few studies have assessed the efficacy of selective HDAC6i in UM or MUM cells in vitro or in vivo. However, evidence in vitro and in pre-clinical UM models suggests inhibiting HDAC6 may offer therapeutic benefit. Nencetti et al. reported that novel compound VS13, with increased HDAC6 selectivity, significantly reduced 92.1 and Mel270 (primary) UM cell viability (up to a 100% reduction) in a dose-dependent manner [
35]. Additionally, ACY-1215, a selective HDAC6i, induced a dose-dependent, significant reduction in UM and MUM cell survival by up to 99.99% in vitro and significantly decreased UM cell fluorescence by 65% in zebrafish xenografts with MUM (OMM2.5) cells in vivo [
36].
Table 1. HDAC6 inhibitors in clinical trials for various cancers.
4. Involvement of HDAC6 in Tumor Growth, Survival and Progression
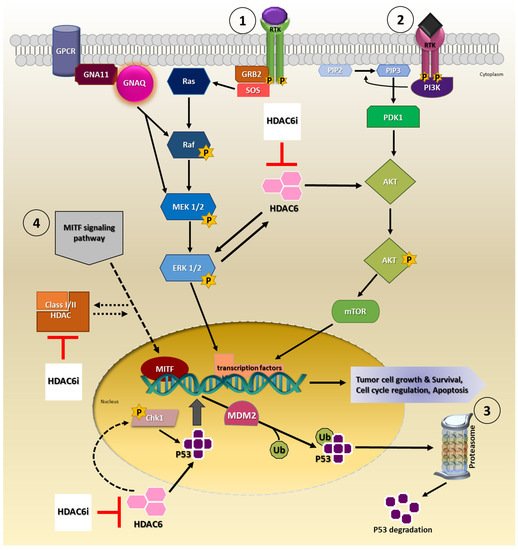
Available evidence indicates HDAC6 is capable of modulating tumor growth, development, survival and progression via signaling pathways such as mitogen-activated protein kinases/extracellular-signal-regulated kinase (MAPK/ERK), phosphatidylinositol 3-kinase (PI3K/AKT) and p53 signaling cascade in cancers (
Figure 1) [
23,
37,
38,
39,
40,
41,
42]. Of these signaling pathways, MAPK/ERK and PI3K/AKT signaling, in particular, are of importance as they are implicated in UM disease pathomechanisms [
43]. In the majority of UM tumors, MAPK/ERK signaling pathway is constitutively activated, which is partly attributed to mutations in genes, guanine nucleotide-binding protein G(q) subunit alpha (
GNAQ) and guanine nucleotide-binding protein G(q) subunit alpha-11 (
GNA11), upstream of this signaling cascade [
44,
45]. Uveal melanoma cells treated with a MEK and PI3K inhibitor combination inhibited cell proliferation and induced apoptosis [
46]. Moreover, several other studies explored MEK and PI3K/AKT pathway inhibitors as a therapeutic option for UM; however, the therapeutic efficacy has been contentious [
43,
47,
48,
49,
50].
Figure 1. Involvement of HDAC6 in cancer signaling pathways. Proposed model for HDAC6i mechanism of action by targeting either ① MAPK/ERK, ② PI3K/AKT, ③ P53 and/or ④ MITF signaling pathway(s). Consequently, targeting these pathways inhibits biological processes that promotes cell survival and proliferation in the cancer cell.
Studies show modulation of HDAC6 in HEK293T cells, either by siRNA mediated knock-down or ACY-1215 pharmacological inhibition, led to increased phospho-ERK levels and ERK1/2 activation, respectively, hence regulating ERK signaling [
51,
52]. In LNCaP prostate cancer cells, treatment with Panobinostat inhibited HDAC6 activity and triggered ERK activation, which consequently resulted in the arrest of cell cycle [
51]. In colorectal cancer cells, HCT116 and HT29, knock-down of HDAC6, blocked cell proliferation, migration and invasion in part via the MAPK/ERK signaling pathway [
53]. A reduction in the expression levels of phospho-MEK, phospho-ERK and phospho-AKT was demonstrated in these cells. Peng et al. (2017), reported that inhibition of cell proliferation and survival with ACY-1215 and/or Vemurafenib in A375 melanoma cells was partly mediated through the inhibition of ERK activation [
54]. Similarly, it was observed that ACY-1215 treatment of esophageal squamous cell carcinoma cells, EC109 and TE-1, led to a decrease in phospho-ERK1/2, phospho-AKT levels and inhibited cell proliferation and survival [
55].
The PI3K/AKT signaling pathway is constitutively activated in UM which facilitates tumorigenesis processes such as cell survival, inhibition of apoptosis and angiogenesis [
56]. In ACY-1215 treated cholangiocarcinoma cells, cell proliferation was blocked, and apoptosis was triggered via the PI3K/AKT pathway [
57]. Kaliszczak et al. (2016) demonstrated that cotreatment of HCT116 with HDAC6i and pan-AKT inhibitor/dual PI3K/mTOR inhibitor enhanced anti-tumor effects both in vitro and in vivo [
58]. Other studies reveal that dual inhibition with HDACi and/or PI3K/AKT/mTOR inhibitor effectively reduced tumor cell proliferation in cancer cells, e.g., prostate cancer, multiple myeloma, relapsed or refractory diffuse large B-cell lymphoma, neuroblastoma, hematologic tumor(s), hepatocarcinoma and breast cancer cells [
59,
60,
61,
62,
63].
Another significant pathway involved in UM disease pathogenesis may be attributed to the p53 signaling pathway. Mutation(s) in
TP53 is rare in UM tumors; however, its activity is commonly disrupted as a consequence of mutations and dysregulation of other key factors in this pathway [
64,
65]. Cao et al. demonstrated in vitro and in vivo, that ACY-1215 increased the transcriptional activity of p53, which consequently led to cell cycle arrest and apoptosis in triple-negative breast cancer cells [
66]. In colorectal cancer cells, HDAC6 inhibition upregulated p53 levels and increased acetylated p53 levels, and consequently increasing apoptosis [
67]. In another study, the authors proposed that in ovarian cancer cells, HDAC6 inhibition in combination with Paclitaxel activated p53 and induced apoptosis [
40]. Recently, it was reported that ACY-1215 inhibited cell proliferation through the promotion of apoptosis through mitotic catastrophe in a p53-dependent manner by repressing p-Chk1 activity, in head and neck carcinoma cells [
68]. Promisingly, a Class III specific HDAC inhibitor, Tenovin-6, prevented tumor cell growth and induced apoptosis by increasing p53 expression in uveal melanoma tumor and cancer stem cells [
69].
Interestingly, an additional pathway receiving renewed attention is the microphthalmia-associated transcription factor (MITF) signaling pathway in UM. MITF signaling is of particular interest in the context of UM as mutations in this transcription factor and its interacting partners are associated with oncogenic functions and disease pathogenesis in cutaneous melanoma [
70,
71,
72,
73]. MITF is widely known as the master regulator of melanogenesis and melanocyte differentiation, in addition to regulating various cellular processes [
74]. Comparably, in UM, MITF seems to have an intricate role balancing between UM tumor growth and tumor suppression activities [
75]. Most recently, Phelps et al. presented that MITF acts as a bona fide tumor suppressor in UM [
76]. The authors report using zebrafish models that loss of
mitfa resulted in growth of UM tumors. Yokoyama et al. (2008) has also shown that pan-HDACi repressed M-MITF expression in clear cell sarcoma and melanoma cells [
77]. Regardless, the exact relationship between HDACi/HDAC6i and MITF still remains to be determined.
As such these pathways offer additional novel putative targets that needs to be thoroughly investigated. Taken together, there is a strong possibility that using HDAC6i may offer therapeutic benefits mediated through the regulation of these highlighted pathways either as a monotherapy or as combinatorial therapy for UM, advanced UM and/or other solid tumors, which needs to be explored extensively.
5. Immunomodulatory Effects of HDAC6 Inhibitors
The tumor microenvironment (TME) supports processes involved in tumorigenesis [
78,
79]. Composed of tumor cells, immune cells, stromal cells, signaling molecules, extracellular matrix and blood vessels, the microenvironment can ensure tumor cells are able to grow, survive, spread and even to gain resistance to therapy [
79]. An increasing number of studies are focusing on understanding and identifying novel therapeutic targets within the tumor microenvironment for cancer treatment. In UM, tumors with chromosomal defects such as loss of one copy of chromosome 3 or gain of chromosome 8q, present with increased levels of inflammatory mediators and immune cells leading to a tumor-promoting inflammatory TME [
80]. Differing from other cancers, increased amounts of tumor infiltrating lymphocytes (TILs) and tumor associated macrophages (TAMs) are correlated to poor prognosis and high metastasis risk in UM [
81,
82]. A delicate balance is required to ensure the prevention of UM cells from evading immune surveillance.
HDACi are capable of immunomodulation in cancer [
38,
83,
84,
85,
86]. Inhibition of Class I HDACs by HDACi or in combination with the immunomodulatory drug Lenalidomide, resulted in the downregulation of cellular Myc proto-oncogene protein (c-MYC) and increased cytotoxicity in multiple myeloma cells [
87]. Additionally, ACY-1215 treatment, alone or combined with Lenalidomide, significantly reduced c-MYC, IKAROS family zinc finger (IKZF)1/IKZF3 and interferon regulatory factor 4 (IRF4) expression levels triggering immune system activation, which was postulated to be involved in the anti-tumor cell survival effects. The HDAC6 inhibitor A452 in combination with Lenalidomide or Pomalidomide, displayed significantly increased synergistic anti-proliferative effects which was attributed to the augmented reduction of c-MYC, IKZF1/IKZF3 and IRF4 expression in multiple myeloma cells [
88]. In a preclinical mouse model of non-small cell lung cancer (NSCLC), ACY-1215 treatment increased the expression of MHC Class II molecules, CD86 and CD96 co-stimulatory molecules, suggesting that ACY-1215 may play a role in T-cell activation and antigen presentation, consequently promoting anti-tumor immunity [
89]. Another study described that the HDAC6 inhibitor ACY-241 alone, and when combined with Oxaliplatin (chemotherapy drug), promoted T cell functions, thereby increasing the immunogenicity of tumor cells, in an NSCLC mouse model [
90]. Inhibition of HDAC6 in melanoma cells resulted in the increased expression of MHC class I and presentation of tumor-related antigens [
91]. Knox et al. demonstrated that treatment of murine melanoma model with the HDAC6i Nexturastat A, combined with the anti-PD1 antibody, increased tumor infiltration of CD8
+ and natural killer cells and reduced the level of pro-tumorigenic M2 macrophages [
92]. Combining HDAC6i with immunomodulatory agents can improve therapeutic efficacy [
93].
9. Conclusion
A combined therapeutic approach involving HDAC6i with chemotherapeutic agents, may be more beneficial and should be explored in detail as treatment options for UM/MUM. Promising results from Entinostat pembrolizumab gives us hope that it is worthwhile to pursue combinatorial treatment strategy in UM/MUM.