Emerging data suggest that chromic hyperinsulinemia is also a driving force for increased activation of the hypothalamic-adrenal-pituitary (HPA) axis in subjects with the metabolic syndrome, leading to a state of “functional hypercortisolism”. This “functional hypercortisolism” by antagonizing insulin actions may prevent hypoglycemia. It also disturbs energy balance by shifting energy fluxes away from muscles toward abdominal fat stores. Synergistic effects of hyperinsulinemia and “functional hypercortisolism” promote abdominal visceral obesity and insulin resistance which are core pathophysiological components of the metabolic syndrome. It is hypothesized that hyperinsulinemia-induced increased activation of the HPA axis plays an important etiological role in the development of the metabolic syndrome and its consequences. Numerous studies have demonstrated reversibility of hyperinsulinemia with lifestyle, surgical, and pharmaceutical-based therapies. Longitudinal studies should be performed to investigate whether strategies that reduce hyperinsulinemia at an early stage are successfully in preventing increased activation of the HPA axis and the metabolic syndrome.
1. Introduction
In the Western populations there is a cluster of metabolic risk factors which include hypertension, glucose intolerance, abdominal obesity, and hyperlipidemia. Since these factors are observed together more frequently than by chance alone, the concept was developed that these factors are interrelated and together produce the metabolic syndrome
[1]. The metabolic syndrome increases risks for developing cardiovascular diseases (such as heart attacks and strokes), type 2 diabetes mellitus, and cancer. The metabolic syndrome has its own set of underlying risk factors of which hyperinsulinemia and insulin resistance, defined as a subnormal response (of blood glucose levels) to insulin, are considered the most important. Other risk factors for the metabolic syndrome are abdominal adiposity, physical inactivity, aging, stress, sleep disturbance, circadian disruption, and the Western diet.
For many years, the dogma has been that insulin resistance was the primary etiological factor in the development of obesity, the metabolic syndrome, and type 2 diabetes, and preceded hyperinsulinemia
[2][3]: hyperinsulinemia was considered secondary representing a compensatory mechanism to overcome systemic (peripheral) insulin resistance. However, at present there is no satisfactory explanation for how insulin resistance might stimulate insulin secretion, while there are only a few naturally occurring or genetic models of primary insulin resistance, and few diabetes genes are implicated in insulin resistance
[3].
The direct contributions of insulin itself in causing or sustaining insulin resistance have received little sustained attention
[2]. Recent data place hyperinsulinemia mechanistically upstream of insulin resistance and suggest that insulin hypersecretion, rather than beta cell dysfunction, identifies otherwise normal individuals at risk for type 2 diabetes
[4]. After a period of 3 years, Ferrannini et al. found in a prospective study that subjects with higher insulin secretion at baseline were more likely to progress to impaired glucose tolerance or T2D than those with lower insulin secretion
[5]. This result, which is consistent with other human studies
[6][7][8][9][10], suggests thus that primary insulin hypersecretion is a triggering event in T2D pathogenesis
[11][12] (
Figure 1). In this alternate concept, hyperinsulinemia precedes impaired insulin secretory function during the conversion from normoglycemia to (pre)diabetes onset: hyperinsulinemia-induced insulin resistance is considered a physiological protective defense mechanism of the body that tries to prevent hypoglycemia and to protect critical tissues from metabolic stress and nutrient-induced injury
[12][13][14]. Thus, hyperinsulinemia may be both a result and a driver of insulin resistance
[2]. In this latter case, hyperinsulinemia may be the direct consequence of consumption of the “modern” Western diet, overnutrition, decreased hepatic insulin clearance, genetic factors, fetal/metabolic programming, defects in pancreatic β-cells, and loss of pulsatile insulin secretion
[12] (
Figure 2).
Figure 1. (
A) The traditional model vs. the alternate model of the pathogenesis of insulin resistance. The traditional model (which is a widely held view) posits that insulin resistance leads to hyperinsulinemia, which in time is followed by β-cell dysfunction (
left). In the traditional model, insulin resistance (1) precedes hyperinsulinemia (2), which may be followed by β-cell exhaustion and finally frank type 2 diabetes. (
B) In the alternate model, hypersecretion of insulin and the resulting hyperinsulinemia (1) primarily cause insulin resistance (2), which may be followed by β-cell exhaustion and finally frank type 2 diabetes (
right). Note that in the alternate model hyperinsulinemia is already present when there is still a normal glucose tolerance. Reproduced from
[12].
Figure 2. Factors involved in primary hyperinsulinemia. Consumption of the “modern” Western diet, over-nutrition, genes, defects in pancreatic β-cells, decreased hepatic insulin clearance, loss of pulsatile insulin secretion, and fetal/metabolic programming may increase insulin secretion, thereby causing chronic hyperinsulinemia and hyperinsulinemia-induced insulin resistance. In the alternate concept, hyperinsulinemia precedes impaired insulin secretory function during the conversion from normoglycemia to (pre) diabetes onset: hyperinsulinemia-induced insulin resistance is considered a physiological protective defense mechanism of the body that tries to prevent hypoglycemia and to protect critical tissues from metabolic stress and nutrient-induced injury.
The metabolic syndrome and Cushing syndrome share several features, including abdominal visceral obesity, insulin resistance, impaired glucose tolerance, and type 2 diabetes mellitus, hypertension, and hypertriglyceridemia (
Table 1). The overlap in clinical characteristics between the metabolic syndrome and Cushing syndrome may be caused by common underlying mechanisms. Earlier studies did not see consistent relationships between plasma cortisol levels and the presence of the metabolic syndrome
[15]. However, new data suggest (a mild) hyperactivity of the HPA axis inducing a state of “functional hypercortisolism” in subjects with the metabolic syndrome
[16][17][18]. Despite the hyperactivity of the HPA axis, in most subjects with the metabolic syndrome, plasma cortisol concentrations are usually low/normal, while simultaneously urinary free cortisol clearance is increased
[16][17][18] (
Table 1). It has been suggested that increased cortisol clearance is responsible for the observed low plasma cortisol concentrations in subjects with the metabolic syndrome. With decreased negative feedback at the hypothalamus and the pituitary, low plasma cortisol might in turn result in an enhanced adrenocorticotropic hormone (ACTH)-induced cortisol response
[19]. An alternative explanation for the low/normal plasma cortisol concentrations in subjects with the metabolic syndrome might be hyperreactivity of the target cells to cortisol
[20].
Table 1. Overlap and differences in clinical characteristics between the metabolic syndrome and Cushing syndrome.
| |
Metabolic Syndrome |
Cushing
Syndrome |
| Plasma Insulin levels |
↑↑ |
↑ * or ↓ ** |
| insulin resistance |
+ |
+ |
| AbdominalObesity |
+ |
+ |
| impaired glucose tolerance |
+ |
+ |
| Hypertriglyceridemia |
+ |
+ |
| Hypertension |
+ |
+ |
| HPA Activity |
↑ |
↑ |
| Plasma Cortisol |
↓/N |
↑ |
| 24 h-Free Cortisoluria |
↑ |
↑ |
Subjects with endogenous Cushing syndrome and the metabolic syndrome differ significantly with respect to insulin secretion. While hyperinsulinemia in the metabolic syndrome is primary, hyperinsulinemia in endogenous Cushing syndrome is secondary, representing a compensatory mechanism to overcome insulin resistance. Moreover, while hyperinsulinemia is an obligatory finding in subjects with the metabolic syndrome, subjects with endogenous hypercortisolemia (Cushing syndrome) and impaired glucose tolerance show a relative hypoinsulinemia, wherein insulin levels have increased, but less than would be expected for the level of plasma glucose
[21]. In subjects with Cushing syndrome as the endogenous hypercortisolemia exacerbates, the relative insulinopenia becomes more paramount, suggesting that relative insulinopenia is caused by cortisol-mediated direct or indirect “toxic” effects on the pancreatic β-cells, which suppresses endogenous insulin secretion
[21][22]. As a direct consequence, insulin secretion in subjects with Cushing syndrome will not suffice to adequately control plasma glucose in response to a glucose load
[21][22][23] (
Table 1). Excess cortisol has negative effects on insulin secretion, insulin sensitivity, and glucose tolerance but does not primarily induce hyperinsulinemia (see below paragraph
Effects of glucocorticoids on insulin secretion for more details).
2. Regulation of the Hypothalamic-Pituitary-Adrenal (HPA)-Axis in Healthy Subjects
Hypothalamus and pituitary regulate cortisol synthesis and release in healthy subjects (
Figure 3). Corticotropin-releasing hormone (CRH) is released by the hypothalamus and stimulates the anterior pituitary to release ACTH. ACTH then acts on the adrenal cortex which promotes the secretion of cortisol from the zona fasciculata. The secretion of cortisol provides a negative feedback loop by inhibiting release of CRH and ACTH from the hypothalamus and anterior pituitary, respectively. The activity of 11 beta-hydroxysteroid dehydrogenase (11b-HSD) plays an important role in extra-adrenal cortisol metabolism
[24]. At least two isozymes of 11 beta-HSD exist, which catalyze the interconversion of hormonally active glucocorticoids (cortisol, corticosterone) and their inactive metabolites (cortisone, 11-dehydrocorticosterone)
[25] 11β-hydroxysteroid dehydrogenase-2 (11β-HSD2), is mainly expressed in the kidneys and protects the mineralocorticoid receptor from glucocorticoid excess by converting cortisol to cortisone
[25]. It thereby promotes the access of aldosterone to the mineralocorticoid receptors in the kidney. The other isoform, 11β-hydroxysteroid dehydrogenase-1 (11β-HSD1), is widely expressed in classic insulin target tissues as the liver, muscle, and adipose tissue.
Figure 3. In healthy subjects, corticotropin-releasing hormone (CRH) stimulates the secretion of adrenocorticotropic hormone (ACTH) in the anterior lobe of the pituitary gland. ACTH in turn acts on the adrenal cortex which produces glucocorticoid hormones (mainly cortisol in humans) in response to stimulation by ACTH. Cortisol in turn acts back on the hypothalamus and the pituitary (to suppress CRH and ACTH) production in a negative feedback cycle. Stimulatory effects are shown in green and inhibitory effects are shown in red. See text for more details.
Circulating levels of cortisol are regulated through a balance between synthesis in the adrenal cortex and clearance via metabolic pathways in the liver. An increase in the level of circulating cortisol may be related to stimulation of 11β-HSD type 1 in the liver and/or reduced cortisol clearance by inhibition of 5α- reductase or 5β-reductase
[24].
Cortisol metabolism is not only regulated centrally, but is also regulated peripherally. A rise of 11β-HSD1 activity (locally) at the tissue level stimulates conversion of cortisone to cortisol, and in this way cortisol receptors are locally exposed to increased cortisol concentrations
[26][27]. Cortisol itself stimulates 11β-HSD1 expression in hepatocytes, adipocytes, and myoblasts
[18]. Moreover, other factors which may increase 11β-HSD1 expression in a tissue-specific manner are tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), and interleukin-6 (IL-6), whereas insulin, insulin-like growth factor-I (IGF-I), and growth hormone reduce 11β-HSD1 activity
[18].
Visceral and omental adipocytes show a higher number of cortisol receptors than subcutaneous adipocytes
[28]. In addition, 11β-HSD1 in visceral and omental adipocytes converts locally more cortisone to (active) cortisol than subcutaneous adipocytes
[29]. As an immediate result, visceral and omental adipocytes are exposed to higher cortisol concentrations than are present in the circulation and this may contribute to the preferential deposition of fat at intra-abdominal rather than subcutaneous sites
[28][30].
3. The Bidirectional Interactions between Insulin and the HPA Axis
In healthy subjects, insulin normally shows a reciprocal relationship with cortisol: insulin inhibits food intake while cortisol stimulates food intake
[31]. Insulin and cortisol are major antagonistic regulators of energy balance. Effects of cortisol and insulin on food intake may be mediated through regulation of hypothalamic neuropeptide-Y (NPY) synthesis and secretion
[31]. In the arcuate nuclei, insulin inhibits and cortisol stimulates the expression of NPY mRNA, which may explain in part the reciprocal actions of insulin and the HPA axis on energy acquisition during the day. It has been further suggested that inhibition of insulin transport across the blood brain barrier by glucocorticoids could be the basis for the enhanced appetite seen with glucocorticoid treatments
[32].Chronic exposure to high circulating glucocorticoid levels inhibits insulin release by binding to glucocorticoid receptors present on pancreatic beta cells
[23].Chronic exposure to glucocorticoids may further reduce the insulinotropic effects of glucagon-like peptide-1 (GLP-1)
[33]. Thus, with several mechanisms, glucocorticoids may inhibit pancreatic β-cell mediated insulin secretion and thereby induce a “relative hypoinsulinemia”. In addition, glucocorticoids may induce insulin resistance (see next paragraph
Glucocorticoids, insulin resistance and metabolism). As long as pancreatic insulin secretion is sufficient, cortisol-mediated increase of insulin resistance and hepatic glucose production will not materially affect glucose tolerance. However, failure of the pancreas to mount an adequate compensatory insulinemic response (due to cortisol-mediated suppression of pancreatic insulin release) may lead to hyperglycemia and impaired glucose tolerance (
Figure 4).
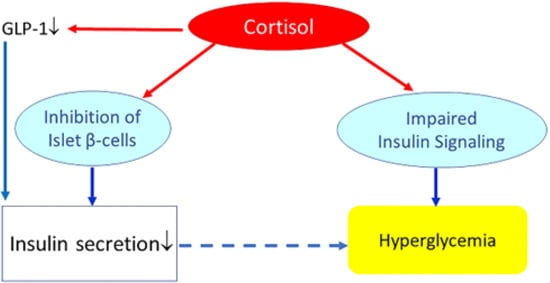
Figure 4. Effects of (high) glucocorticoids on insulin, insulin resistance, and glucose metabolism. Cortisol directly suppresses pancreatic insulin release. It also reduces glucagon-like peptide-1 (GLP-1) production, which further decreases insulin secretion. Increased cortisol also induces glycogenolysis and the expression of key gluconeogenic enzymes, which will increase hepatic glucose production and release. In addition, cortisol may impair insulin receptor signaling (at the receptor and post-receptor level) and thereby induce insulin resistance. Failure of the pancreas to mount an adequate compensatory insulinemic response (“relative hypoinsulinemia” due to cortisol-mediated suppression of pancreatic insulin release) with cortisol-induced insulin resistance may lead to hyperglycemia and impaired glucose tolerance. However, if insulin secretion is sufficient to overcome cortisol-mediated insulin resistance, cortisol will not materially affect glucose tolerance.
4. Hyperinsulinemia Induces a State of “Functional Hypercortisolism”
As previously discussed, numerous recent data are supportive of the concept that hyperinsulinemia per se is primary and causes insulin resistance
[3][11] (
Figure 1). In this alternate concept, insulin resistance is proposed to be a physiological defense mechanism of the body preventing hyperinsulinemia-induced hypoglycemia and protecting against overstimulation of target tissues from metabolic stress and nutrient-induced injury
[12][13][14]. This concept is even more interesting against emerging data suggesting that chromic hyperinsulinemia is also a driving force for increased activation of the hypothalamic-adrenal-pituitary (HPA) axis in subjects with the metabolic syndrome, leading to a state of “functional hypercortisolism”. Thus hyperinsulinemia-induced increased activation of the HPA axis plays an important etiological role in the development of the metabolic syndrome and its consequences. At this moment it is unclear whether there is a successful strategy to modify hyperinsulinemia-induced “functional hypercortisolism” in subjects prone to develop the metabolic syndrome.
5. Conclusions
In conclusion, emerging data suggest that chromic hyperinsulinemia is the driving force for increased HPA axis activity in subjects with the metabolic syndrome. This “functional hypercortisolism” by antagonizing insulin actions may prevent hypoglycemia, disturb energy homeostasis, and shift energy fluxes away from muscle toward fat stores. Synergistic effects of hyperinsulinemia and “functional hypercortisolism” promote fat accumulation in visceral fat cells and so contribute to abdominal obesity. Chronic hyperinsulinemia-induced activation of the HPA axis may play an important etiological role in the development of the metabolic syndrome and all its consequences.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23158178


