Mitochondria are not only the main energy supplier but are also the cell metabolic center regulating multiple key metaborates that play pivotal roles in epigenetics regulation. These metabolites include acetyl-CoA, α-ketoglutarate (α-KG), S-adenosyl methionine (SAM), NAD+, and O-linked beta-N-acetylglucosamine (O-GlcNAc), which are the main substrates for DNA methylation and histone post-translation modifications, essential for gene transcriptional regulation and cell fate determination. Tumorigenesis is attributed to many factors, including gene mutations and tumor microenvironment. Mitochondria and epigenetics play essential roles in tumor initiation, evolution, metastasis, and recurrence. Targeting mitochondrial metabolism and epigenetics are promising therapeutic strategies for tumor treatment.
- mitochondria
- epigenetics
- cancer
1. Introduction
2. Mitochondria Function in Epigenetic Regulation
2.1. Mitochondria and DNA Methylation
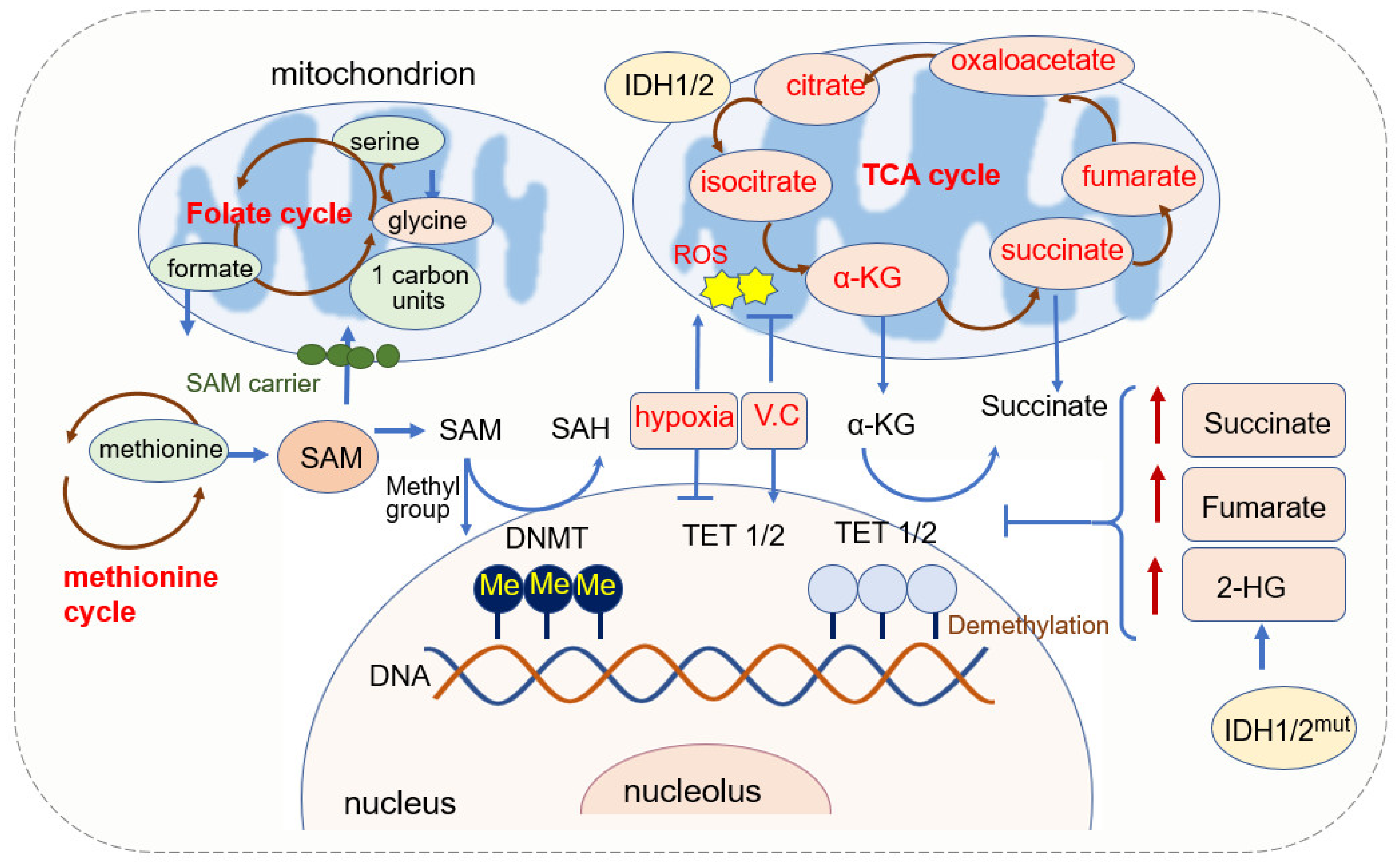
2.1.1. SAM Source and Mitochondrial Regulation of SAM
2.1.2. α-KG Source and Its Function in DNA Methylation
2.1.3. Mitochondrial ROS and DNA Methylation
2.1.4. Mitochondria Dysfunction and DNA Methylation
2.2. Mitochondria and RNA Methylation
2.2.1. The Effect of Mitochondria-Associated Metabolites on RNA Methylation
2.2.2. The Roles of RNA Methylation in Mitochondria
This entry is adapted from the peer-reviewed paper 10.3390/cells11162518
References
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934.
- Archibald, J.M. Endosymbiosis and Eukaryotic Cell Evolution. Curr. Biol. 2015, 25, R911–R921.
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159.
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698.
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638.
- Janssen, J.J.E.; Grefte, S.; Keijer, J.; de Boer, V.C.J. Mito-Nuclear Communication by Mitochondrial Metabolites and Its Regulation by B-Vitamins. Front. Physiol. 2019, 10, 78.
- Rigon, M.; Townley, A.R.; Campanella, M. Mitochondria ensure immune surveillance by retro-communication with the nucleus. Cell Metab. 2021, 33, 853–855.
- Scatena, R. Mitochondria and drugs. Adv. Exp. Med. Biol. 2012, 942, 329–346.
- Liu, Y.E.; Shi, Y.F. Mitochondria as a target in cancer treatment. Medcomm 2020, 1, 129–139.
- Chen, K.; Zhang, J.; Beeraka, N.M.; Tang, C.; Babayeva, Y.V.; Sinelnikov, M.Y.; Zhang, X.; Zhang, J.; Liu, J.; Reshetov, I.V.; et al. Advances in the Prevention and Treatment of Obesity-Driven Effects in Breast Cancers. Front. Oncol. 2022, 12, 820968.
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2022, 83, 556–569.
- Matilainen, O.; Quiros, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463.
- Waddington, C.H. The Epigenotype. Int. J. Epidemiol. 2011, 41, 10–13.
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232.
- Riggs, A.D. X-Inactivation, Differentiation, and DNA Methylation. Cytogenet. Genome Res. 1975, 14, 9–25.
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2021, 13, 877–919.
- Daniel, M.; Tollefsbol, T.O. Epigenetic linkage of aging, cancer and nutrition. J. Exp. Biol. 2015, 218, 59–70.
- Martin, S.L.; Hardy, T.M.; Tollefsbol, T.O. Medicinal chemistry of the epigenetic diet and caloric restriction. Curr. Med. Chem. 2013, 20, 4050–4059.
- Ross, S.A.; Dwyer, J.; Umar, A.; Kagan, J.; Verma, M.; Van Bemmel, D.M.; Dunn, B.K. Introduction: Diet, epigenetic events and cancer prevention. Nutr. Rev. 2008, 66 (Suppl. S1), S1–S6.
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31.
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55.
- Bowden, G.T.; Schneider, B.; Domann, R.; Kulesz-Martin, M. Oncogene activation and tumor suppressor gene inactivation during multistage mouse skin carcinogenesis. Cancer Res. 1994, 54, 1882s–1885s.
- Solomon, H.; Brosh, R.; Buganim, Y.; Rotter, V. Inactivation of the p53 tumor suppressor gene and activation of the Ras oncogene: Cooperative events in tumorigenesis. Discov. Med. 2010, 9, 448–454.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970.
- Blackadar, C.B. Historical review of the causes of cancer. World J. Clin. Oncol. 2016, 7, 54–86.
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540.
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280.
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648.
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134.
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401.
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611.
- Zhu, L.Q.; Gibson, P.; Currle, D.S.; Tong, Y.; Richardson, R.J.; Bayazitov, I.T.; Poppleton, H.; Zakharenko, S.; Ellison, D.W.; Gilbertson, R.J. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 2009, 457, 603–607.
- Roskams, T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene 2006, 25, 3818–3822.
- Toh, T.B.; Lim, J.J.; Chow, E.K. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29.
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482.
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Silva Santos, R.D.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176.
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432.
- Cai, S.F.; Levine, R.L. Genetic and epigenetic determinants of AML pathogenesis. Semin. Hematol. 2019, 56, 84–89.
- Ilango, S.; Paital, B.; Jayachandran, P.; Padma, P.R.; Nirmaladevi, R. Epigenetic alterations in cancer. Front. Biosci. 2020, 25, 1058–1109.
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral Heterogeneity of the Epigenome. Cancer Cell 2016, 29, 440–451.
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386.
- Murphy, S.K.; Jirtle, R.L. Imprinting evolution and the price of silence. Bioessays 2003, 25, 577–588.
- Das, R.; Hampton, D.D.; Jirtle, R.L. Imprinting evolution and human health. Mamm. Genome 2009, 20, 563–572.
- Esteller, M. Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum. Mol. Genet. 2007, 16, R50–R59.
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334.
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892.
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829.
- Peng, Y.; Liu, H.; Liu, J.; Long, J. Post-translational modifications on mitochondrial metabolic enzymes in cancer. Free Radic. Biol. Med. 2022, 179, 11–23.
- Dai, Z.; Ramesh, V.; Locasale, J.W. The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 2020, 21, 737–753.
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450.
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18.
- Margueron, R.; Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296.
- Xu, S.; Zhang, X.; Liu, C.; Liu, Q.; Chai, H.; Luo, Y.; Li, S. Role of Mitochondria in Neurodegenerative Diseases: From an Epigenetic Perspective. Front. Cell Dev. Biol. 2021, 9, 688789.
- Paik, W.K.; Paik, D.C.; Kim, S. Historical review: The field of protein methylation. Trends Biochem. Sci. 2007, 32, 146–152.
- Bird, A.P. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980, 8, 1499–1504.
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257.
- Li, C.; Fan, Y.; Li, G.; Xu, X.; Duan, J.; Li, R.; Kang, X.; Ma, X.; Chen, X.; Ke, Y.; et al. DNA methylation reprogramming of functional elements during mammalian embryonic development. Cell Discov. 2018, 4, 41.
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56.
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402.
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92.
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38.
- Wang, J.; Zhang, K.X.; Lu, G.Z.; Zhao, X.H. Research progress on 5hmC and TET dioxygenases in neurodevelopment and neurological diseases. Yi Chuan 2017, 39, 1138–1149.
- Matuleviciute, R.; Cunha, P.P.; Johnson, R.S.; Foskolou, I.P. Oxygen regulation of TET enzymes. FEBS J. 2021, 288, 7143–7161.
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935.
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of alpha-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30.
- Wang, J.; Tang, J.; Lai, M.; Zhang, H. 5-Hydroxymethylcytosine and disease. Mutat. Res. Rev. Mutat. Res. 2014, 762, 167–175.
- Valinluck, V.; Sowers, L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007, 67, 946–950.
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA demethylation dynamics. Cell 2011, 146, 866–872.
- Jin, S.G.; Kadam, S.; Pfeifer, G.P. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010, 38, e125.
- Ouyang, Y.; Wu, Q.; Li, J.; Sun, S.; Sun, S. S-adenosylmethionine: A metabolite critical to the regulation of autophagy. Cell Prolif. 2020, 53, e12891.
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395.
- Agrimi, G.; Di Noia, M.A.; Marobbio, C.M.; Fiermonte, G.; Lasorsa, F.M.; Palmieri, F. Identification of the human mitochondrial S-adenosylmethionine transporter: Bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem. J. 2004, 379, 183–190.
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280.
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237.
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.J.; Gupta, D.; Mattocks, D.; Padilla, P.G.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E.; et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015, 22, 861–873.
- Ducker, G.S.; Chen, L.; Morscher, R.J.; Ghergurovich, J.M.; Esposito, M.; Teng, X.; Kang, Y.; Rabinowitz, J.D. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016, 24, 640–641.
- Martelli, M.P.; Martino, G.; Cardinali, V.; Falini, B.; Martinelli, G.; Cerchione, C. Enasidenib and ivosidenib in AML. Minerva Med. 2020, 111, 411–426.
- Liu, S.; He, L.; Yao, K. The Antioxidative Function of Alpha-Ketoglutarate and Its Applications. Biomed. Res. Int. 2018, 2018, 3408467.
- Baracco, E.E.; Castoldi, F.; Durand, S.; Enot, D.P.; Tadic, J.; Kainz, K.; Madeo, F.; Chery, A.; Izzo, V.; Maiuri, M.C.; et al. alpha-Ketoglutarate inhibits autophagy. Aging 2019, 11, 3418–3431.
- Asadi Shahmirzadi, A.; Edgar, D.; Liao, C.Y.; Hsu, Y.M.; Lucanic, M.; Asadi Shahmirzadi, A.; Wiley, C.D.; Gan, G.; Kim, D.E.; Kasler, H.G.; et al. Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice. Cell Metab. 2020, 32, 447–456.e6.
- Long, L.H.; Halliwell, B. Artefacts in cell culture: Alpha-Ketoglutarate can scavenge hydrogen peroxide generated by ascorbate and epigallocatechin gallate in cell culture media. Biochem. Biophys. Res. Commun. 2011, 406, 20–24.
- Tran, K.A.; Dillingham, C.M.; Sridharan, R. The role of alpha-ketoglutarate-dependent proteins in pluripotency acquisition and maintenance. J. Biol. Chem. 2019, 294, 5408–5419.
- Baksh, S.C.; Finley, L.W.S. Metabolic Coordination of Cell Fate by alpha-Ketoglutarate-Dependent Dioxygenases. Trends Cell Biol. 2021, 31, 24–36.
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69.
- Yang, Q.; Liang, X.; Sun, X.; Zhang, L.; Fu, X.; Rogers, C.J.; Berim, A.; Zhang, S.; Wang, S.; Wang, B.; et al. AMPK/alpha-Ketoglutarate Axis Dynamically Mediates DNA Demethylation in the Prdm16 Promoter and Brown Adipogenesis. Cell Metab. 2016, 24, 542–554.
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388.
- Sciacovelli, M.; Goncalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.H.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.B.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547.
- Xiao, M.T.; Yang, H.; Xu, W.; Ma, S.H.; Lin, H.P.; Zhu, H.G.; Liu, L.X.; Liu, Y.; Yang, C.; Xu, Y.H.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Gene Dev. 2012, 26, 1326–1338.
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G., Jr.; Godley, L.A.; Koivunen, P. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes. J. Biol. Chem. 2016, 291, 4256–4265.
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239.
- Hoekstra, A.S.; de Graaff, M.A.; Briaire-de Bruijn, I.H.; Ras, C.; Seifar, R.M.; van Minderhout, I.; Cornelisse, C.J.; Hogendoorn, P.C.; Breuning, M.H.; Suijker, J.; et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget 2015, 6, 38777–38788.
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744.
- Thienpont, B.; Galle, E.; Lambrechts, D. TET enzymes as oxygen-dependent tumor suppressors: Exciting new avenues for cancer management. Epigenomics 2016, 8, 1445–1448.
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728.
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485.
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species 2016, 1, 9–21.
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive oxygen species (ROS)—Induced genetic and epigenetic alterations in human carcinogenesis. Mutat. Res. 2011, 711, 167–173.
- Quan, X.; Lim, S.O.; Jung, G. Reactive oxygen species downregulate catalase expression via methylation of a CpG island in the Oct-1 promoter. FEBS Lett. 2011, 585, 3436–3441.
- Lim, S.O.; Gu, J.M.; Kim, M.S.; Kim, H.S.; Park, Y.N.; Park, C.K.; Cho, J.W.; Park, Y.M.; Jung, G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: Methylation of the E-cadherin promoter. Gastroenterology 2008, 135, 2128–2140.e8.
- Wu, Q.; Ni, X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr. Drug Targets 2015, 16, 13–19.
- Afanas’ev, I. New nucleophilic mechanisms of ros-dependent epigenetic modifications: Comparison of aging and cancer. Aging Dis. 2014, 5, 52–62.
- Fan, W.; Luo, J.Y. SIRT1 Regulates UV-Induced DNA Repair through Deacetylating XPA. Mol. Cell 2010, 39, 247–258.
- Kang, K.A.; Zhang, R.; Kim, G.Y.; Bae, S.C.; Hyun, J.W. Epigenetic changes induced by oxidative stress in colorectal cancer cells: Methylation of tumor suppressor RUNX3. Tumour Biol. 2012, 33, 403–412.
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91.
- Fuks, F. DNA methylation and histone modifications: Teaming up to silence genes. Curr. Opin. Genet. Dev. 2005, 15, 490–495.
- Delatte, B.; Jeschke, J.; Defrance, M.; Bachman, M.; Creppe, C.; Calonne, E.; Bizet, M.; Deplus, R.; Marroqui, L.; Libin, M.; et al. Genome-wide hydroxymethylcytosine pattern changes in response to oxidative stress. Sci. Rep. 2015, 5, 12714.
- Kriaucionis, S.; Heintz, N. The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science 2009, 324, 929–930.
- Chia, N.; Wang, L.; Lu, X.; Senut, M.C.; Brenner, C.; Ruden, D.M. Hypothesis: Environmental regulation of 5-hydroxymethylcytosine by oxidative stress. Epigenetics 2011, 6, 853–856.
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20.
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674.
- Yin, R.; Mao, S.Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403.
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquiere, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68.
- MacLeod, A.R.; Rouleau, J.; Szyf, M. Regulation of DNA methylation by the Ras signaling pathway. J. Biol. Chem. 1995, 270, 11327–11337.
- Ploumakis, A.; Coleman, M.L. OH, the Places You’ll Go! Hydroxylation, Gene Expression, and Cancer. Mol. Cell 2015, 58, 729–741.
- Singh, K.K. Mitochondria damage checkpoint in apoptosis and genome stability. FEMS Yeast Res. 2004, 5, 127–132.
- Minocherhomji, S.; Tollefsbol, T.O.; Singh, K.K. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 2012, 7, 326–334.
- Smiraglia, D.J.; Kulawiec, M.; Bistulfi, G.L.; Gupta, S.G.; Singh, K.K. A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol. Ther. 2008, 7, 1182–1190.
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282.
- Matsuda, S.; Yasukawa, T.; Sakaguchi, Y.; Ichiyanagi, K.; Unoki, M.; Gotoh, K.; Fukuda, K.; Sasaki, H.; Suzuki, T.; Kang, D. Accurate estimation of 5-methylcytosine in mammalian mitochondrial DNA. Sci. Rep. 2018, 8, 5801.
- Mechta, M.; Ingerslev, L.R.; Fabre, O.; Picard, M.; Barres, R. Evidence Suggesting Absence of Mitochondrial DNA Methylation. Front. Genet. 2017, 8, 166.
- Hong, E.E.; Okitsu, C.Y.; Smith, A.D.; Hsieh, C.L. Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol. Cell Biol. 2013, 33, 2683–2690.
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635.
- Sharma, J.; Kumari, R.; Bhargava, A.; Tiwari, R.; Mishra, P.K. Mitochondrial-induced Epigenetic Modifications: From Biology to Clinical Translation. Curr. Pharm. Des. 2021, 27, 159–176.
- Bicci, I.; Calabrese, C.; Golder, Z.J.; Gomez-Duran, A.; Chinnery, P.F. Single-molecule mitochondrial DNA sequencing shows no evidence of CpG methylation in human cells and tissues. Nucleic Acids Res. 2021, 49, 12757–12768.
- Yang, B.; Wang, J.Q.; Tan, Y.; Yuan, R.; Chen, Z.S.; Zou, C. RNA methylation and cancer treatment. Pharm. Res. 2021, 174, 105937.
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769.
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975.
- Perry, R.P.; Kelley, D.E. Existence of methylated messenger RNA in mouse L cells. Cell 1974, 1, 37–42.
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624.
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206.
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 2012, 149, 1635–1646.
- Deng, X.; Su, R.; Weng, H.; Huang, H.; Li, Z.; Chen, J. RNA N6-methyladenosine modification in cancers: Current status and perspectives. Cell Res. 2018, 28, 507–517.
- Huo, F.C.; Zhu, Z.M.; Pei, D.S. N6-methyladenosine (m6 A) RNA modification in human cancer. Cell Prolif. 2020, 53, e12921.
- Zhang, X.; Wei, L.H.; Wang, Y.X.; Xiao, Y.; Liu, J.; Zhang, W.; Yan, N.; Amu, G.; Tang, X.J.; Zhang, L.; et al. Structural insights into FTO’s catalytic mechanism for the demethylation of multiple RNA substrates. Proc. Natl. Acad. Sci. USA 2019, 116, 2919–2924.
- Liu, Y.; You, Y.; Lu, Z.; Yang, J.; Li, P.; Liu, L.; Xu, H.; Niu, Y.; Cao, X. N6-methyladenosine RNA modification-mediated cellular metabolism rewiring inhibits viral replication. Science 2019, 365, 1171–1176.
- Li, Z.J.; Weng, H.Y.; Su, R.; Weng, X.C.; Zuo, Z.X.; Li, C.Y.; Huang, H.L.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N-6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141.
- Su, R.; Li, Z.J.; Weng, H.Y.; Weng, X.C.; Zuo, Z.X.; Li, C.Y.; Huang, H.L.; Hu, C.; Qin, X.; Tang, L.C.; et al. Fto Plays an Oncogenic Role in Acute Myeloid Leukemia As a N-6-Methyladenosine RNA Demethylase. Blood 2016, 128, 2706.
- Scholler, E.; Marks, J.; Marchand, V.; Bruckmann, A.; Powell, C.A.; Reichold, M.; Mutti, C.D.; Dettmer, K.; Feederle, R.; Huttelmaier, S.; et al. Balancing of mitochondrial translation through METTL8-mediated m3C modification of mitochondrial tRNAs. Mol. Cell 2021, 81, 4810–4825.
- Morscher, R.J.; Ducker, G.S.; Li, S.H.; Mayer, J.A.; Gitai, Z.; Sperl, W.; Rabinowitz, J.D. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 2018, 554, 128–132.
- Delaunay, S.; Pascual, G.; Feng, B.; Klann, K.; Behm, M.; Hotz-Wagenblatt, A.; Richter, K.; Zaoui, K.; Herpel, E.; Munch, C.; et al. Mitochondrial RNA modifications shape metabolic plasticity in metastasis. Nature 2022, 607, 593–603.
- Vilardo, E.; Amman, F.; Toth, U.; Kotter, A.; Helm, M.; Rossmanith, W. Functional characterization of the human tRNA methyltransferases TRMT10A and TRMT10B. Nucleic Acids Res. 2020, 48, 6157–6169.