Several tumor polyamine-suppressing strategies have been developed, as follows. (1) Ornithine decarboxylase (ODC) and S-adenosylmethionine decarboxylase 1 (AMD1) are important for polyamine synthesis. The α-difluoromethylornithine (DFMO), which acts as an irreversible suicide inhibitor of ODC, has been used to prevent and treat multiple cancers, such as pancreatic cancer, gastric cancer, lung carcinoma, neuroblastoma, endometrial cancer, and osteosarcoma. (2) Highly regulated catabolic pathways are utilized to control the intracellular polyamine pool. The modulation of the polyamine catabolic enzyme produces decreasing polyamine content and induces the generation of toxic compounds. (3) Some inhibitors targeting the polyamine transport system (PTS) can hinder polyamine import and antagonize polyamine uptake. (4) Synthetic polyamines, including polyamine analogs and polyamine conjugates, possess anticancer activity against tumor cells.
1. Small Molecules Target Polyamine Metabolism
1.1. Polyamine Analogs
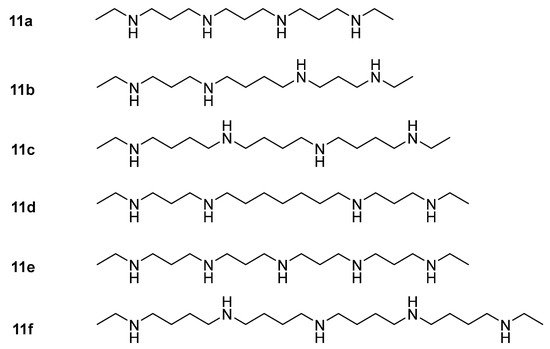
As potential antitumor drugs, polyamine analogs have a similar structure to natural polyamines but cannot be functionally substituted for them. That means that polyamine analogs can compete with natural polyamines, can enter cells through polyamine transport channels, which down-regulates the activities of various polyamine synthases, and can promote polyamine catabolism. In this way, the intracellular content of natural polyamines is depleted by inhibiting biosynthesis and inducing catabolism. For example, propylenediamine-based polyamine analogs and bis(ethyl)spermine analogs (
11a–
11f,
Figure 1) were synthesized to inhibit the ornithine decarboxylase (ODC) activity and growth of tumor cells [
72,
73].
Figure 1. Chemical structures of polyamine analogs
11a–
11f [
73].
1.2. Polyamine Conjugates
Polyamine conjugates are usually composed of polyamine and a DNA intercalator. The former is responsible for driving the conjugates to be transported to cancer cells by polyamine transport system (PTS), competitively inhibiting the uptake of exogenous polyamines by tumor cells. The latter can effectively embed into DNA, destroying many DNA protein interactions and interfering with their function, resulting in cell death. DNA intercalators are capable of intercalating into nucleic acid base pairs. The classic intercalators include naphthalimide, flavonoids, naphthoquinone, anthraquinone, and chalcone.
Some naphthalimide-polyamine conjugates significantly inhibited tumor growth and metastasis [
74,
75,
76]. Xie’s group [
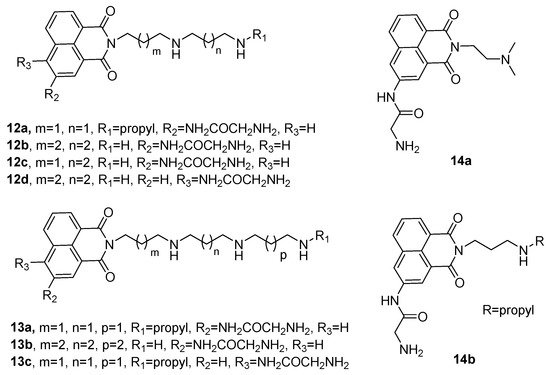
74] designed and synthesized a series of naphthalimide-polyamine conjugates (
12a–
12d,
13a–
13c, and
14a–
14b). They found that compound
13b exhibited stronger antitumor activity against hepatoma cells than the other compounds. Moreover, its activity was significantly higher than that of commercial antitumor drugs (amonafil and cisplatin). The mechanism of action revealed that
13b can regulate the key enzymes of polyamine metabolism with SSAT and PAO and down-regulate the concentrations of spermine, spermidine, and putrescine, thereby inhibiting the rapid growth of the tumor cells (
Figure 2).
Figure 2. Chemical structures of the naphthalimide-polyamine conjugates,
12a–
d,
13a–
c and
14a–
b [
74].
Wang’s group [
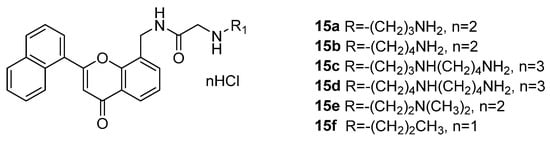
77] reported a series of flavonoid polyamine conjugates (
15a–
15f) and evaluated their anti-tumor properties through in vitro and in vivo experiments (
Figure 3). Overall, the series was moderately cytotoxic to tumor cells. However, only compound
15a showed good selectivity between hepatocellular carcinoma cells (HCC) and normal hepatocytes. When
15a was combined with aspirin, the anti-HCC activity was improved. The H22 liver tumor growth and lung metastasis were effectively inhibited. The combinational effect was found, wherein
15a increased the expression of apoptosis-related proteins and aspirin further amplified this effect. Other flavonoid polyamine conjugates can also initiate autophagy and inhibit the occurrence of apoptosis [
78,
79].
Figure 3. Chemical structures of the flavonoid polyamine conjugates,
15a–
15f [
77].
A major enzyme involved in maintaining DNA topology, 1,4-naphthoquinone is capable of inhibiting DNA human topoisomerase II-α (topo2α) and has been widely used in many cancer-related biological target inhibitors [
80]. Follmer’s group [
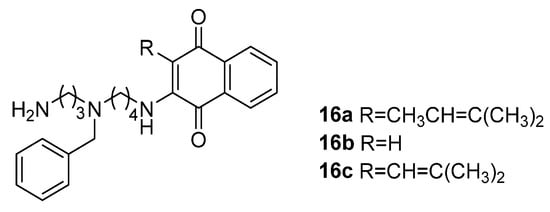
81] constructed a series of 1,4-naphthoquinone-polyamine conjugates (
16a–
16c) to study their anticancer properties (
Figure 4). The coupling of polyamines could enhance the antitumor activity of 1,4-naphthoquinone on tumor cells. Porphyrin-polyamine conjugates [
82], anthraquinone-polyamine conjugates [
83], and chalcone polyamine conjugates [
84] have also been investigated for anti-tumor purposes.
Figure 4. Chemical structures of the naphthoquinone-polyamine conjugates,
16a–
16c [
81].
2. Combinations Target Multiple Components
Due to compensatory mechanisms, the single agents have failed to obtain satisfactory clinical effects. For example, increased polyamine uptake from the surrounding microenvironment is involved if polyamine synthesis is limited. The increased polyamine catabolism leads to an increase in the activity of the ODC polyamine biosynthetic enzyme. When polyamines are depleted in cells, the ODC expression levels are increased. A combination strategy combining two or more agents is more promising for cancer chemoprevention because the agents work synergistically, with decreased adverse effects. For example, “polyamine blocking therapy (PBT)” by a combination of polyamine biosynthesis and transport inhibitors can lead to efficient polyamine depletion. The combination of DFMO and catabolism inducers has shown enhanced antitumor activity.
2.1. Spermine-Responsive Supramolecular Chemotherapy
Supramolecular chemotherapy is devoted to utilizing supramolecular approaches to reduce the cytotoxicity of chemotherapeutic drugs and enhance their anticancer activity. The use of macrocyclic host molecules can encapsulate clinical anticancer drugs and reduce their toxicity to normal cells. Upon entering the polyamine multi-expression environment, the polyamine can competitively combine with the macrocyclic host, thereby causing the release of anti-tumor drug molecules.
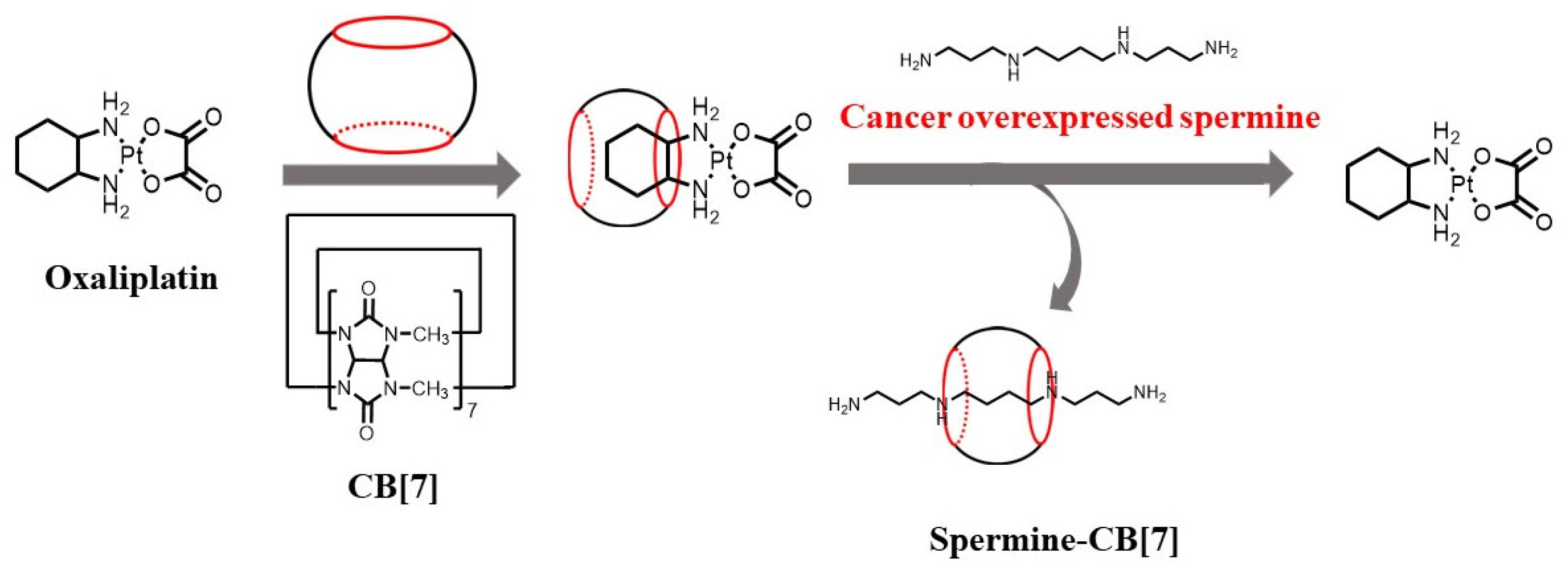
Zhang and co-workers developed supramolecular chemotherapy based on cucurbit[n]uril and an antitumor drug [
85,
86,
87]. Upon competitive replacement by spermine, the recovery of the antitumor activities of drugs was obtained. For example, the host−guest complex of dimethyl viologen (MV)-CB[7] (Cucurbit[7]uril) was added to tumor cells with overexpressed spermine; subsequently, the antitumor activity of MV can be recovered by the competitive replacement of spermine [
85]. When another clinical drug, oxaliplatin, was used as a model antitumor agent, the oxaliplatin-CB[7] complex exhibited more cooperatively enhanced antitumor activity than oxaliplatin itself (
Figure 5) [
86]. The cooperatively enhanced spermine consumption in tumor environments and the release of oxaliplatin were possible reasons.
Figure 5. Supramolecular chemotherapy using oxaliplatin and CB[7] and its competitive release and replacement by spermine [
86].
The supramolecular polymeric chemotherapy results were also promising. PEG-functionalized CB[7] can encapsulate oxaliplatin to form a supramolecular polymeric complex [
87]. Due to the presence of the PEG chain, the improved circulation performance of oxaliplatin was achieved. This combined effect led to enhanced cytotoxicity for tumor cells and decreased cytotoxicity for normal cells.
Supramolecular chitosan nanogels (SCNs) were fabricated, based on phenylalanine-grafted chitosan and cucurbit[8]uril (CB[8]) by stimuli-responsive host−guest interactions [
88]. Doxorubicin hydrochloride (DOX), a chemotherapeutic agent, was entrapped in the matrix to yield DOX-SCNs, with an excellent drug loading efficiency. In the presence of spermine, the encapsulated DOX was selectively released due to a strong host–guest interaction between CB[8] and spermine. The SCNs were efficiently internalized by the cells, and the DOX-SCNs exhibited specific, potent activity against spermine-overexpressed A549 cancerous cells.
Since ZnO can interact with DOX and CB[7], Chen et al. constructed a smart supramolecular cargo of ZnO-DOX-CB[7] via the ion-dipole interaction [
89]. Due to the higher binding affinity between CB[7] and spermine, the nanocomplex could release DOX and enhance antitumor activity through competitive displacement in tumor microenvironments. Similarly, a heptaplatin-CB[7] supramolecular chemotherapy platform was constructed via host−guest interaction for killing tumor cells [
90], since overexpressed spermine competitively exchanged heptaplatin from the supramolecular platform, leading to a stronger anti-proliferative ability than heptaplatin itself.
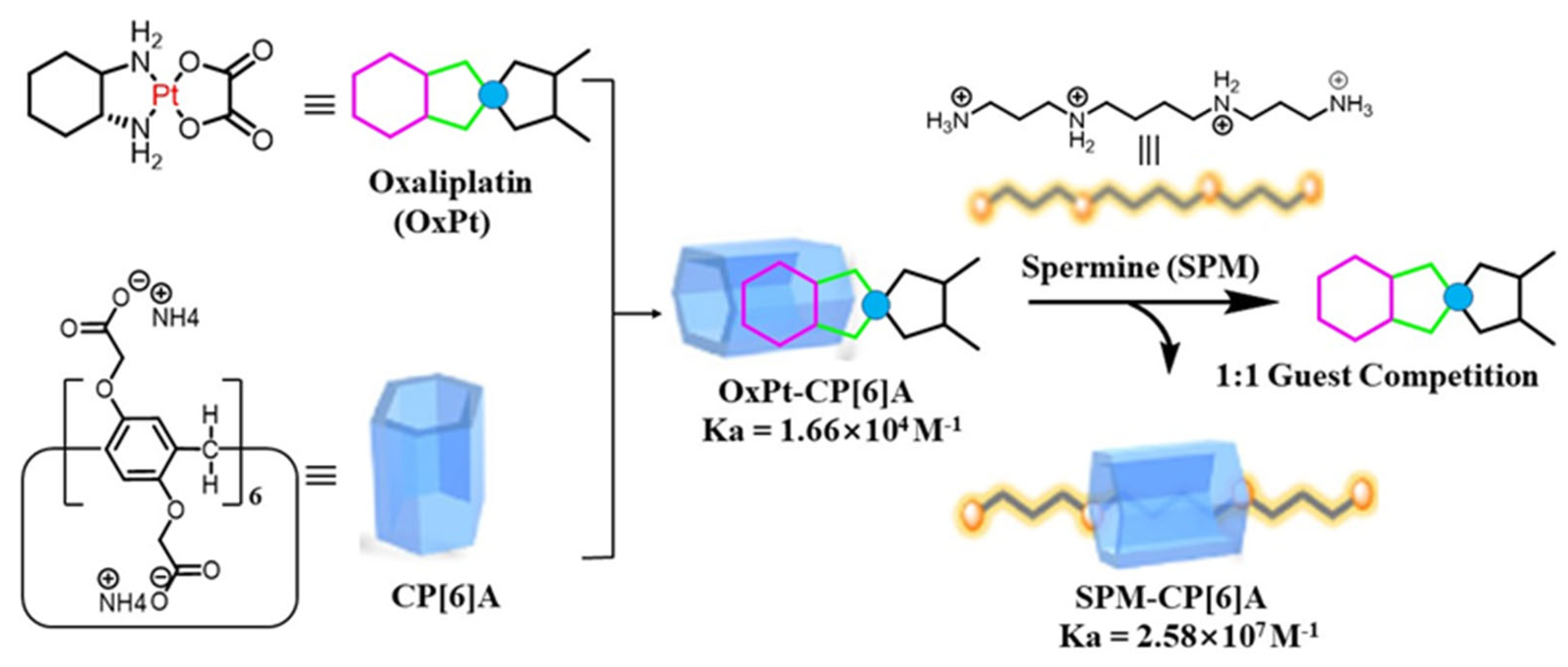
Pillar[n]arene is another popular macrocyclic host in supramolecular chemotherapy. The pillar[n]arene has a high affinity with spermine. Zhang’s group reported that the carboxylated pillar[6]arene-oxaliplatin complex showed a 20% higher anticancer bioactivity than oxaliplatin itself (
Figure 6) [
91]. The possible mechanism was ascribed to encapsulated oxaliplatin that was released from the carboxylated pillar[6]arene-oxaliplatin complex by competitive replacement with spermine. At the same time, the cytotoxicity toward normal cells was reduced.
Figure 6. Supramolecular chemotherapy, based on host–guest complexation between carboxylated pillar[6]arene and oxaliplatin [
91].
Wang’s group reported bispillar[5]arene-paclitaxel nanoparticles that were spermine and glutathione stimuli-responsive, allowing precisely selective drug release in lung cancer cells that overexpress spermine and glutathione [
92]. Peptide-drug conjugates show excellent biocompatibility and tunable morphologies. Wang’s group developed the supramolecular-peptide based on CB[7] and peptide-camptothecin conjugates by non-covalent interactions [
93]. When the supramolecular-peptide was internalized into spermine-overexpressed cancer cells, the free peptide-camptothecin conjugates were released by the competitive binding of spermine with CB[7]. As a result, the drastic morphological transformation from nanoparticles to microfibers led to an improved accumulation and retention of supramolecular peptides in tumor cells. The polyamine-responsive “nanoparticle-to-microfiber” transformation exhibited a potential tumor therapy effect. Li’s group synthesized a peptide-pillar[5]arene conjugate as a supramolecular trap, showing a wide spectrum of antitumor activities [
94].
2.2. Combination of Polyamine Consumption and Photodynamic Therapy (PDT)
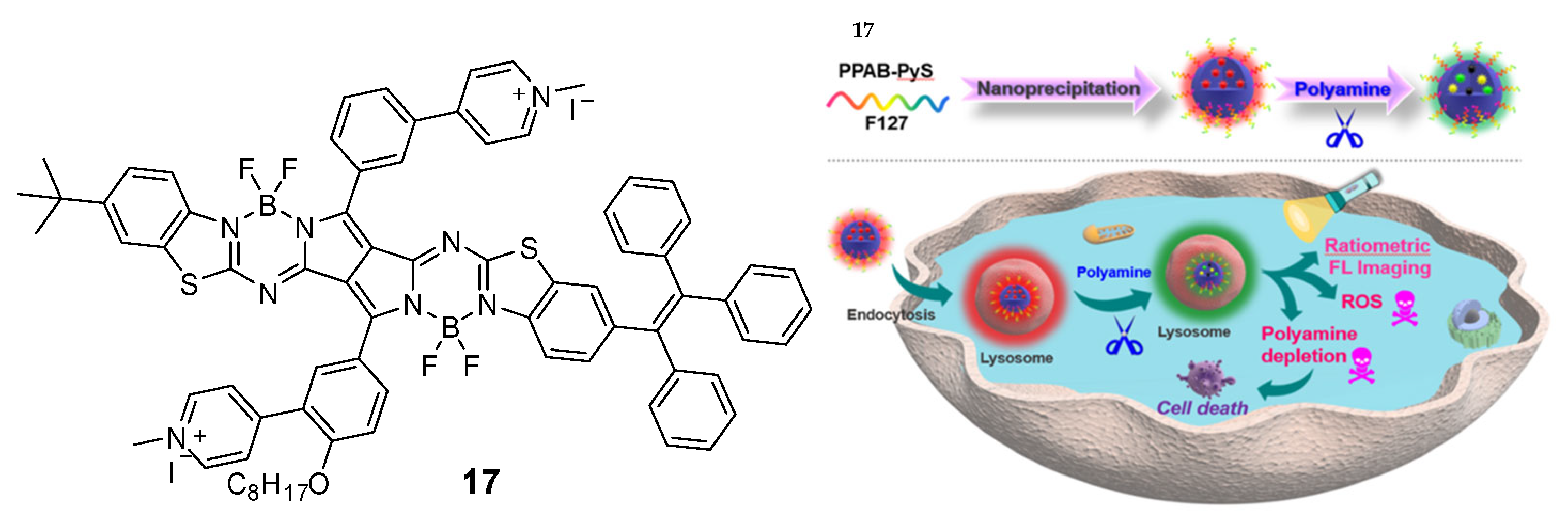
One group explored a new type of PPAB-based photosensitizer (compound
17), which can cooperatively consume polyamine and produce two photosensitizers through an irreversible chemical reaction [
95]. The combination of polyamine consumption and PDT by synergistically destroying tumor cells was a promising polyamine consumption strategy (
Figure 7). Compound
17 had lysosomal targeting ability, ratiometric fluorescence imaging capability, polyamine depletion, and enhanced reactive oxygen species (ROS) generation. Their study provides an irreversible polyamine consumption strategy, which improves anticancer efficacy after being combined with PDT.
Figure 7. The combination of photodynamic therapy with a putative depletion of polyamines on cancer treatment with compound
17 [
95].
This entry is adapted from the peer-reviewed paper 10.3390/bios12080633