The effectiveness of many anticancer drugs depends on the creation of specific metabolites that may alter their therapeutic or toxic properties. One significant route of biotransformation is a conjugation of electrophilic compounds with reduced glutathione, which can be non-enzymatic and/or catalyzed by glutathione-dependent enzymes. Glutathione usually combines with anticancer drugs and/or their metabolites to form more polar and water-soluble glutathione S-conjugates, readily excreted outside the body. In this regard, glutathione plays a role in detoxification, decreasing the likelihood that a xenobiotic will react with cellular targets.
- anticancer drugs
- glutathione
- mechanisms of glutathione conjugation reaction
- detoxification
- bioactivation
1. Introduction: The Place of Glutathione in Drug Metabolism
2. Glutathione
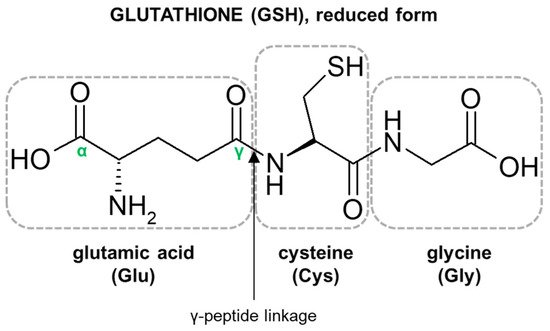
2.1. Glutathione Structure as a Determinant of Its Biological Functions
2.2. Biological Functions of Glutathione
3. Overview of Mechanisms of Glutathione Conjugation of Anticancer Drugs
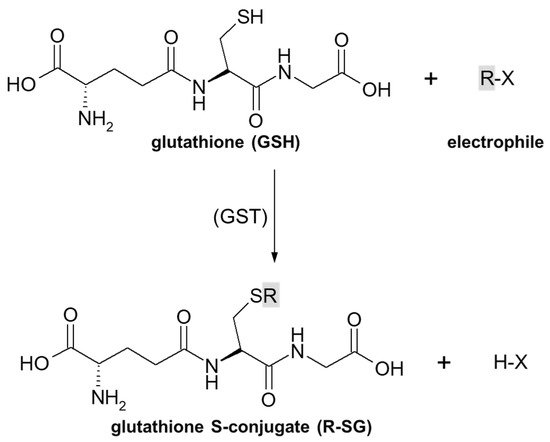
3.1. Nucleophilic Substitution
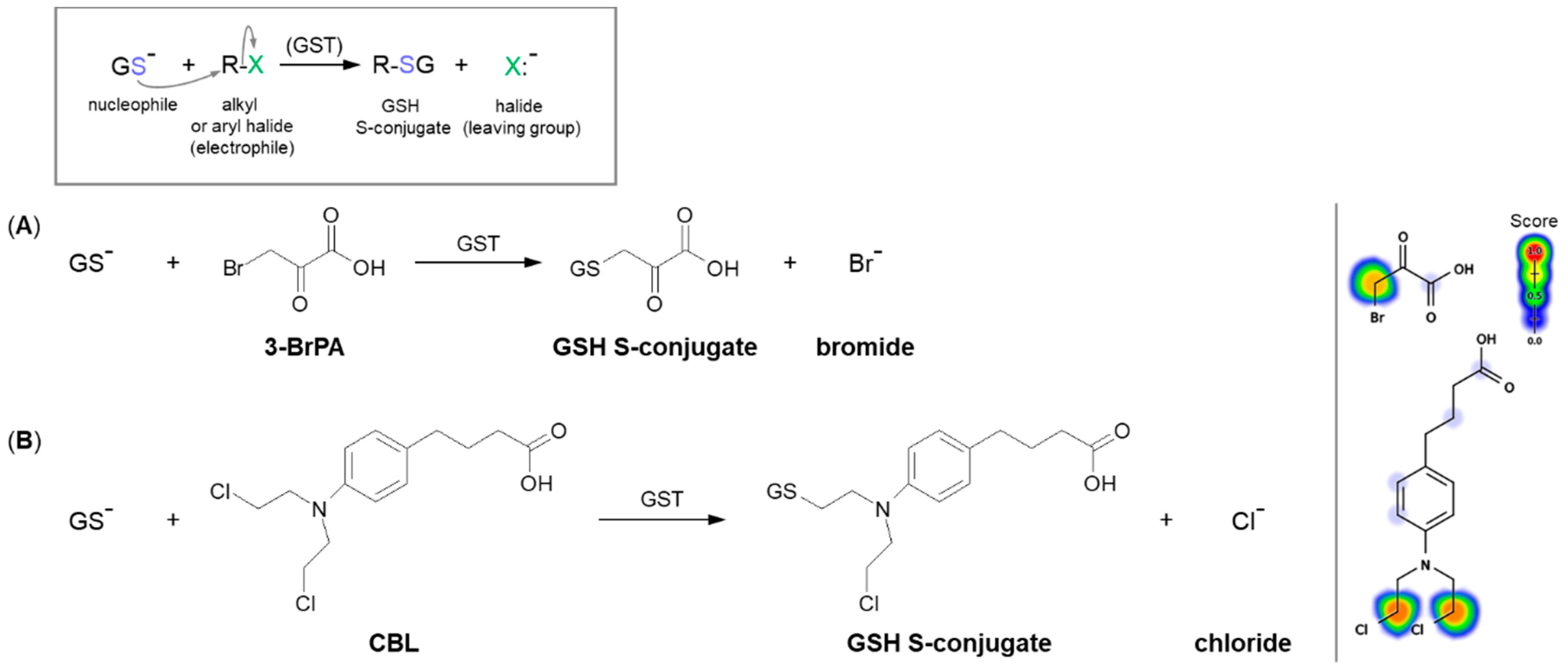
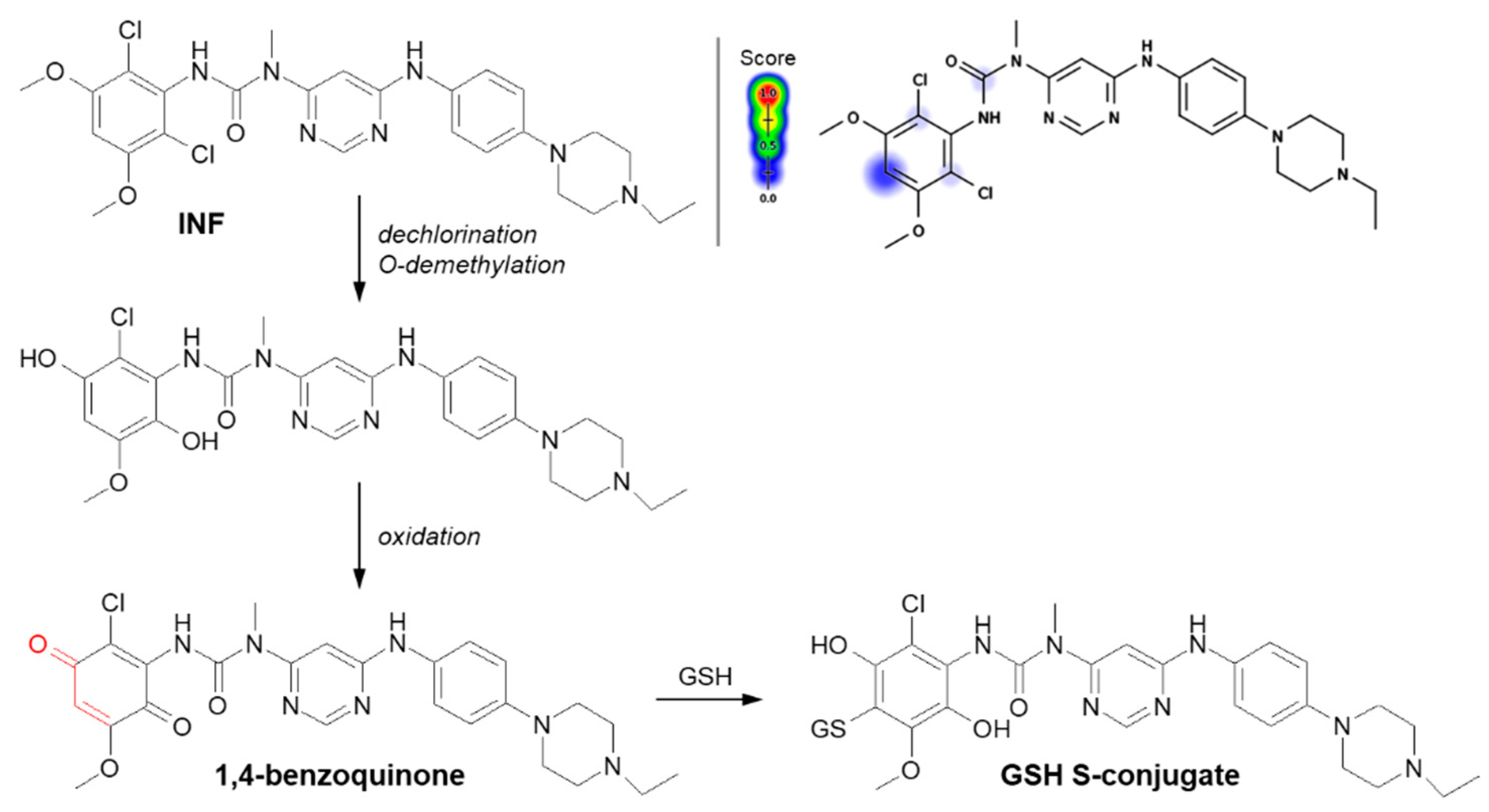
3.1.1. Halogen Atom as a Leaving Group
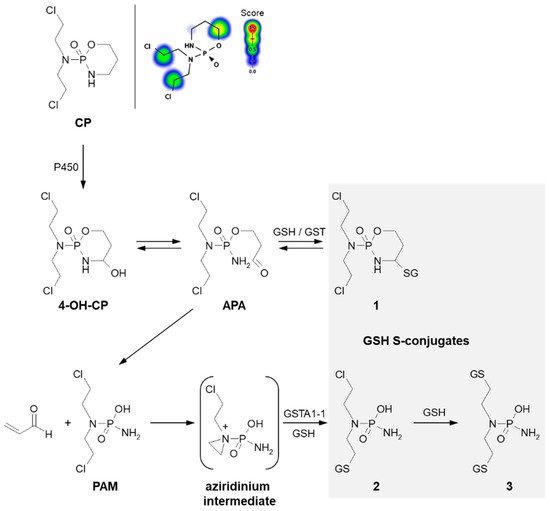
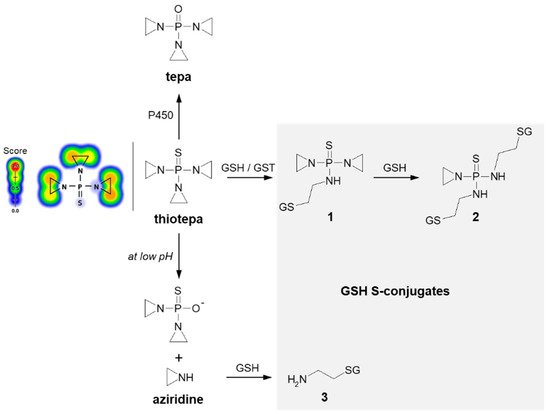
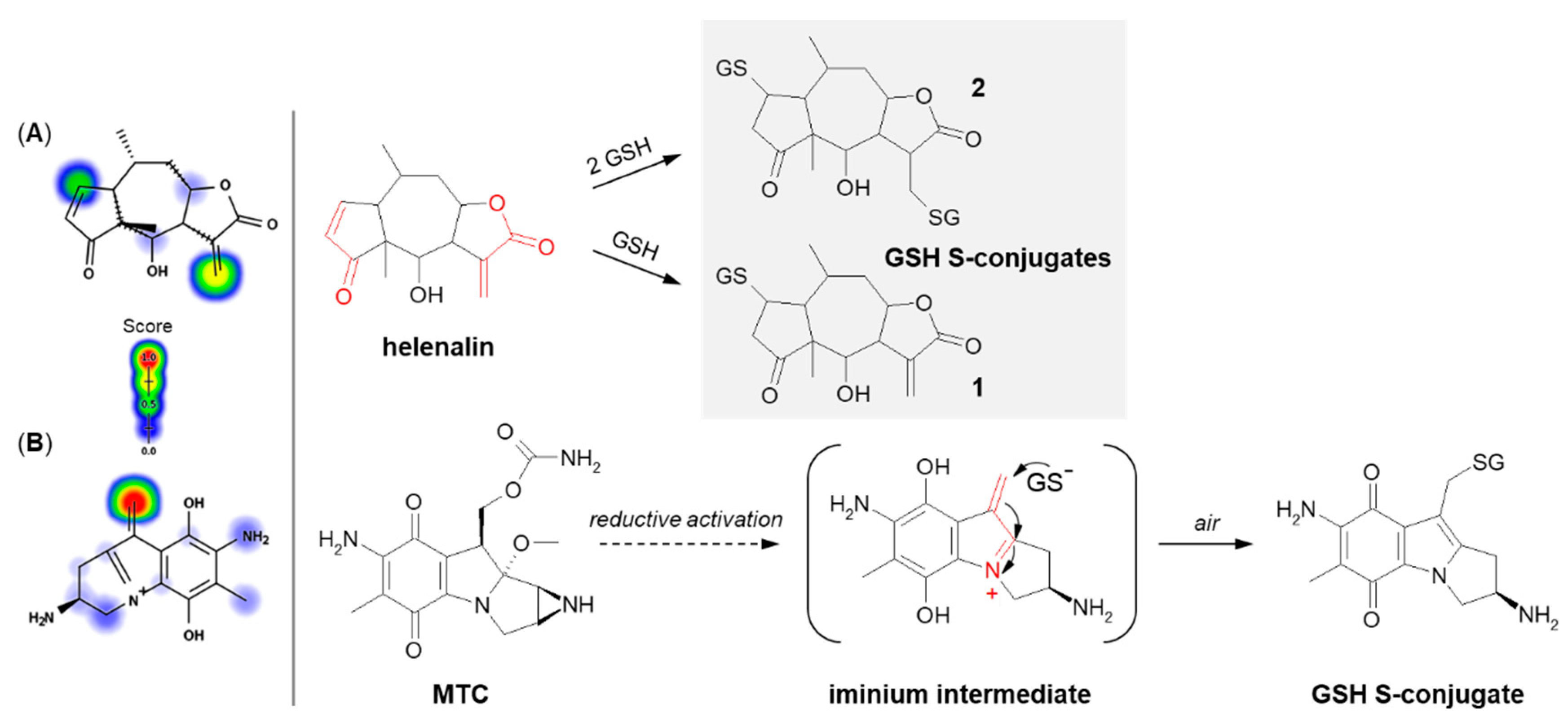
3.1.2. Tensioned Ring-Opening Reaction
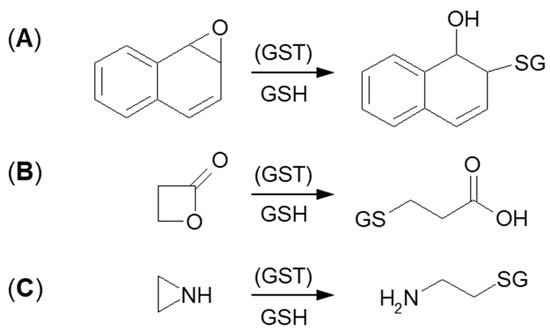
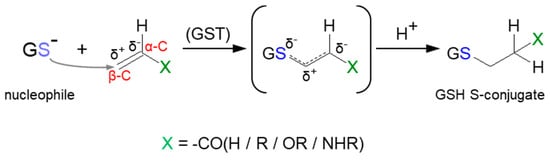
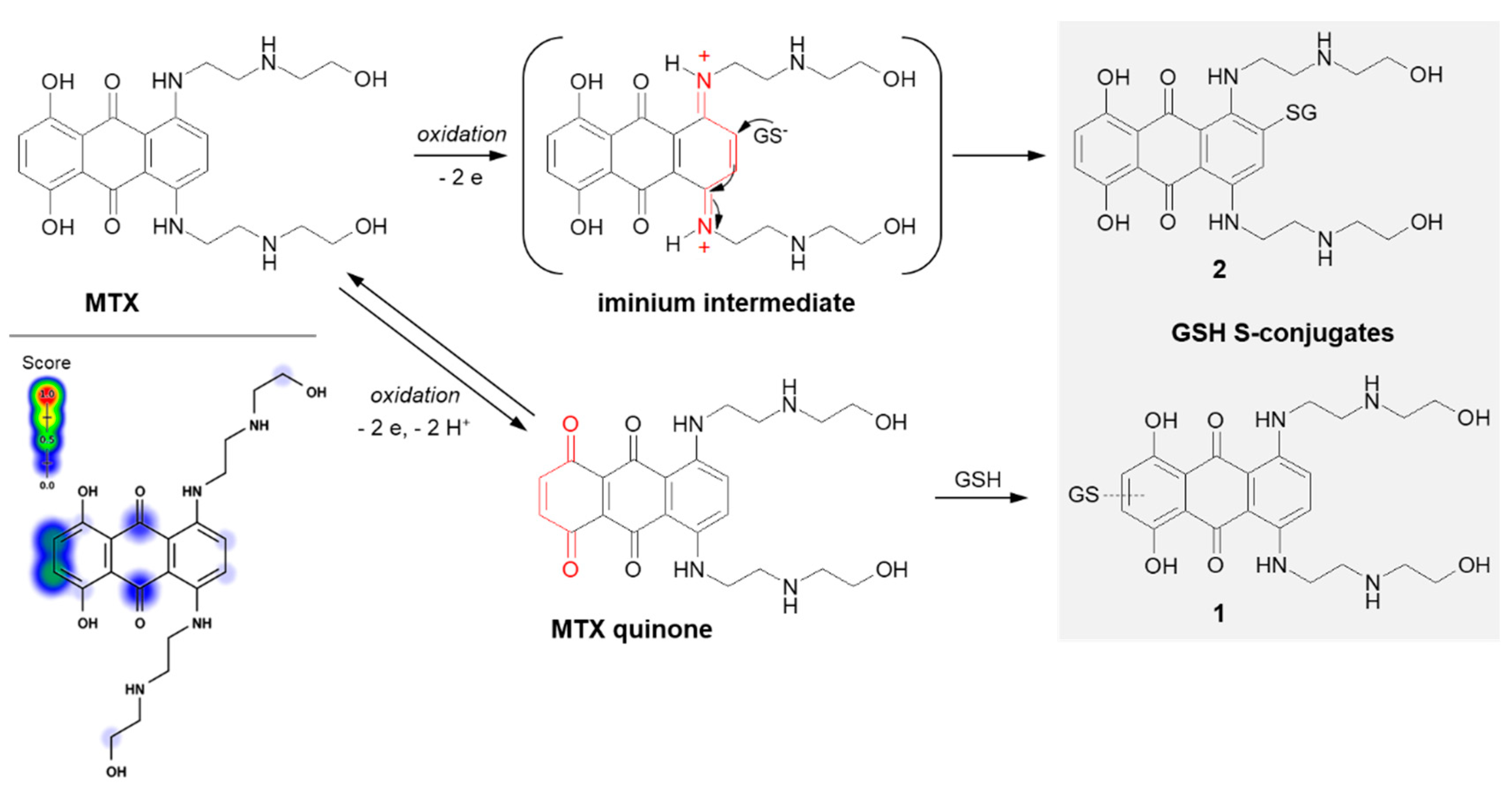
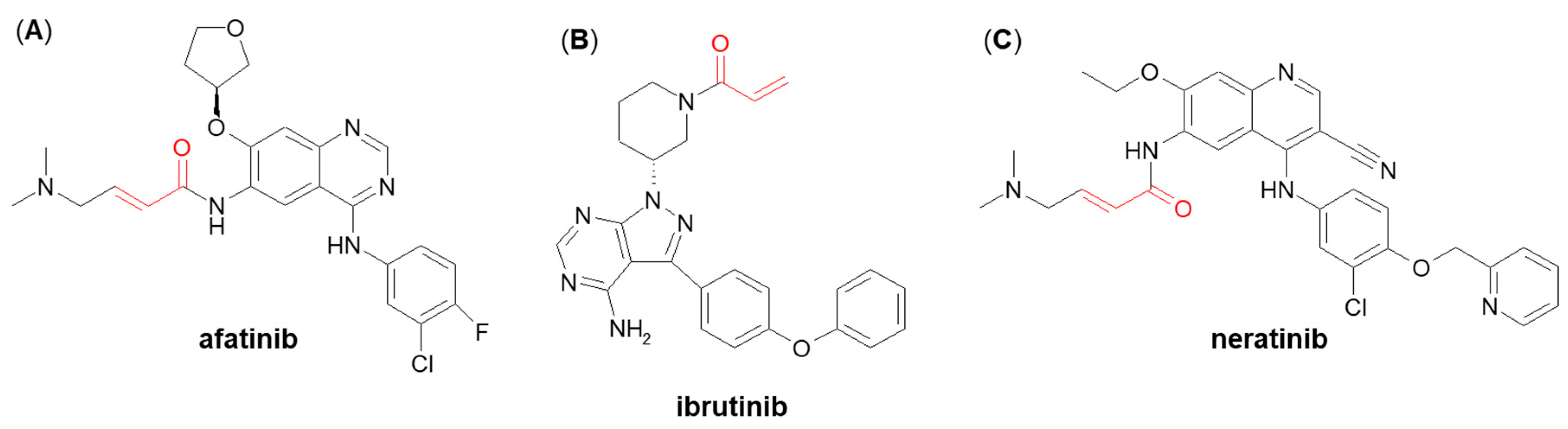
3.2. Nucleophilic Addition (Michael Addition)
This entry is adapted from the peer-reviewed paper 10.3390/molecules27165252
References
- Kumar, G.N.; Surapaneni, S. Role of drug metabolism in drug discovery and development. Med. Res. Rev. 2001, 21, 397–411.
- Bachmann, K. Chapter 8—Drug Metabolism. In Pharmacology: Principles and Practise, 1st ed.; Hacker, M., Messer, W., Bachmann, K., Eds.; Academic Press: San Diego, CA, USA, 2009; pp. 131–173.
- Penner, N.; Woodward, C.; Prakash, C. Drug Metabolizing Enzymes and Biotransformation Reactions. In ADME-Enabling Technologies in Drug Design and Development, 1st ed.; Zhang, D., Surapaneni, S., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 545–565.
- Baillie, T.A. Cytochrome P450 and Other Enzymes in Drug Discovery and Development. In Drug Metabolizing Enzymes, 1st ed.; Lee, J., Obach, R.S., Fisher, M.B., Eds.; CRC Press, LLC: Boca Raton, FL, USA, 2003; pp. 147–154.
- Jančová, P.; Šiller, M. Phase II Drug Metabolism. In Topics on Drug Metabolism; Paxton, J., Ed.; IntechOpen: London, UK, 2012; pp. 35–60.
- Döring, B.; Petzinger, E. Phase 0 and phase III transport in various organs: Combined concept of phases in xenobiotic transport and metabolism. Drug Metab. Rev. 2014, 46, 261–282.
- Attia, S.M. Deleterious effects of reactive metabolites. Oxidative Med. Cell. Longev. 2010, 3, 238–253.
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837.
- Satoh, K. The high non-enzymatic conjugation rates of some glutathione S-transferase (GST) substrates at high glutathione concentrations. Carcinogenesis 1995, 16, 869–874.
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta Gen. Sub. 2013, 1830, 3217–3266.
- Armstrong, R.N. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 1997, 10, 2–18.
- Dirven, H.A.A.M.; Megens, L.; Oudshoorn, M.J.; Dingemanse, M.A.; Van Ommen, B.; Van Bladeren, P.J. Glutathione conjugation of the cytostatic drug ifosfamide and the role of human glutathione S-transferases. Chem. Res. Toxicol. 1995, 8, 979–986.
- Dirven, H.A.A.M.; Van Ommen, B.; Van Bladeren, P.J. Glutathione conjugation of alkylating cytostatic drugs with a nitrogen mustard group and the role of glutathione S-transferases. Chem. Res. Toxicol. 1996, 9, 351–360.
- Cnubben, N.H.; Rommens, A.J.; Oudshoorn, M.J.; Van Bladeren, P.J. Glutathione-dependent biotransformation of the alkylating drug thiotepa and transport of its metabolite monoglutathionylthiotepa in human MCF-7 breast cancer cells. Cancer Res. 1998, 58, 4616–4623.
- Zhang, K.; Wong, K.P.; Chow, P. Conjugation of chlorambucil with GSH by GST purified from human colon adenocarcinoma cells and its inhibition by plant polyphenols. Life Sci. 2003, 72, 2629–2640.
- Rossato, L.G.; Costa, V.M.; De Pinho, P.G.; Arbo, M.D.; De Freitas, V.; Vilain, L.; De Lourdes Bastos, M.; Palmeira, C.; Remião, F. The metabolic profile of mitoxantrone and its relation with mitoxantrone-induced cardiotoxicity. Arch. Toxicol. 2013, 87, 1809–1820.
- El Sayed, S.M.; Baghdadi, H.; Zolaly, M.; Almaramhy, H.H.; Ayat, M.; Donki, J.G. The promising anticancer drug 3-bromopyruvate is metabolized through glutathione conjugation which affects chemoresistance and clinical practice: An evidence-based view. Med. Hypotheses 2017, 100, 67–77.
- Cooper, A.J.L.; Hanigan, M.H. 10.17—Metabolism of Glutathione S-Conjugates: Multiple Pathways. In Comprehensive Toxicology, 3rd ed.; McQueen, C.A., Ed.; Elsevier Ltd.: Oxford, UK, 2018; pp. 363–406.
- Van Bladeren, P.J. Glutathione conjugation as a bioactivation reaction. Chem. Biol. Interact. 2000, 129, 61–76.
- Dekant, W. The Role of Biotransformation and Bioactivation in Toxicity. In Molecular, Clinical and Environmental Toxicology; Luch, A., Ed.; Experientia Supplementum: Basel, Switzerland, 2009; pp. 57–86.
- Konopa, J.K.; Horowska, B.; Paluszkiewicz, E.M.; Borowa-Mazgaj, B.; Augustin, E.A.; Skwarska, A.; Mazerska, Z. Asymmetric Bis-Acridines with Antitumour Activity and Use Thereof. European Patent EP3070078A1, 4 October 2017.
- Konopa, J.K.; Horowska, B.; Paluszkiewicz, E.M.; Borowa-Mazgaj, B.; Augustin, E.A.; Skwarska, A.; Mazerska, Z. Asymmetric Bis-Acridines with Antitumour Activity and Their Uses. United. States Patent US10202349B2, 12 February 2019.
- Kosno, M.; Laskowski, T.; Frackowiak, J.E.; Potęga, A.; Kurdyn, A.; Andrałojć, W.; Borzyszkowska-Bukowska, J.; Szwarc-Karabyka, K.; Mazerska, Z. Acid–base equilibrium and self-association in relation to high antitumor activity of selected unsymmetrical bisacridines established by extensive chemometric analysis. Molecules 2022, 27, 3995.
- Mieszkowska, A.; Nowicka, A.M.; Kowalczyk, A.; Potęga, A.; Pawłowska, M.; Kosno, M.; Augustin, E.; Mazerska, Z. Metabolic Profiles of new unsymmetrical bisacridine antitumor agents in electrochemical and enzymatic noncellular systems and in tumor cells. Pharmaceuticals 2021, 14, 317.
- Potęga, A.; Kosno, M.; Mazerska, Z. Novel insights into conjugation of antitumor-active unsymmetrical bisacridine C-2028 with glutathione: Characteristics of non-enzymatic and glutathione S-transferase-mediated reactions. J. Pharm. Anal. 2021, 11, 791–798.
- Paluszkiewicz, E.; Horowska, B.; Borowa-Mazgaj, B.; Peszyńska-Sularz, G.; Paradziej-Łukowicz, J.; Augustin, E.; Konopa, J.; Mazerska, Z. Design, synthesis and high antitumor potential of new unsymmetrical bisacridine derivatives towards human solid tumors, specifically pancreatic cancers and their unique ability to stabilize DNA G-quadruplexes. Eur. J. Med. Chem. 2020, 204, 11259.
- Chasseaud, L.F. The Role of Glutathione and Glutathione S-Transferases in the Metabolism of Chemical Carcinogens and Other Electrophilic Agents. In Advances in Cancer Research; Klein, G., Weinhouse, S., Eds.; Academic Press: San Diego, CA, USA, 1979; pp. 175–274.
- Anderson, M.E. Glutathione: An overview of biosynthesis and modulation. Chem. Biol. Interact. 1998, 111–112, 1–14.
- Lenton, K.J.; Therriault, H.; Wagner, J.R. Analysis of glutathione and glutathione disulfide in whole cells and mitochondria by postcolumn derivatization high-performance liquid chromatography with ortho-phthalaldehyde. Anal. Biochem. 1999, 274, 125–130.
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208.
- Josephy, P.D.; Mannervik, B. Biochemistry of Glutathione. In Molecular Toxicology, 2nd ed.; Oxford University Press, Inc.: New York, NY, USA, 2006; pp. 333–364.
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214.
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155.
- Ketterer, B.; Coles, B.; Meyer, D.J. The role of glutathione in detoxication. Environ. Health Perspect. 1983, 49, 59–69.
- Park, H.-A.; Khanna, S.; Rink, C.; Gnyawali, S.; Roy, S.; Sen, C.K. Glutathione disulfide induces neural cell death via a 12-lipoxygenase pathway. Cell Death Differ. 2009, 16, 1167–1179.
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762.
- Jeffery, E.H. 2—Biochemical Basis of Toxicity. In Handbook of Toxicologic Pathology, 2nd ed.; Haschek, W.M., Rousseaux, C.G., Wallig, M.A., Eds.; Academic Press: San Diego, CA, USA, 2002; pp. 15–37.
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free Radic. Biol. Med. 2019, 140, 14–27.
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-Glutathionylation: From molecular mechanisms to health outcomes. Antioxid. Redox Signal. 2011, 15, 233–270.
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921.
- Lu, S.C. Regulation of glutathione synthesis. Mol. Aspects. Med. 2009, 30, 42–59.
- Gomez, L.D.; Noctor, G.; Knight, M.R.; Foyer, C.H. Regulation of calcium signalling and gene expression by glutathione. J. Exp. Bot. 2004, 55, 1851–1859.
- Circu, M.L.; Aw, T.J. Glutathione and apoptosis. Free Radic. Res. 2009, 42, 689–706.
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12.
- Arrick, B.A.; Nathan, C.F. Glutathione metabolism as a determinant of therapeutic efficacy: A review. Cancer Res. 1984, 44, 4224–4232.
- Cooper, A.J.L.; Hanigan, M.H. 4.17—Enzymes Involved in Processing Glutathione Conjugates. In Comprehensive Toxicology, 2nd ed.; McQueen, C.A., Ed.; Elsevier Ltd.: Oxford, UK, 2010; pp. 323–366.
- Li, J.C.; Renslo, A. Chapter 6: Nucleophilic Substitution, Addition, and Elimination Reactions. In The Organic Chemistry of Medicinal Agents; Renslo, A., Ed.; McGraw Hill: New York, NY, USA, 2016.
- Ouellette, R.J.; Rawn, J.D. Nucleophilic Substitutions and Elimination Reactions. In Organic Chemistry: Structure, Mechanism, Synthesis, 2nd ed.; Ouellette, R.J., Rawn, J.D., Eds.; Academic Press: San Diego, CA, USA, 2018; pp. 299–319.
- Karpusas, M.; Axarli, I.; Chiniadis, L.; Papakyriakou, A.; Bethanis, K.; Scopelitou, K.; Clonis, Y.D.; Labrou, N.E. The interaction of the chemotherapeutic drug chlorambucil with human glutathione transferase A1-1: Kinetic and structural analysis. PLoS ONE 2013, 8, e56337.
- Azevedo-Silva, J.; Queiros, O.; Baltazar, F.; Ułaszewski, S.; Goffeau, A.; Ko, Y.H.; Pedersen, P.L.; Preto, A.; Casal, M. The anticancer agent 3-bromopyruvate: A simple but powerful molecule taken from the lab to the bedside. J. Bioenerg. Biomembr. 2016, 48, 349–362.
- El Sayed, S.M.; Mohamed, W.G.; Seddik, M.-A.H.; Ahmed, A.-S.A.; Mahmoud, A.G.; Amer, W.H.; Helmy Nabo, M.M.; Hamed, A.R.; Ahmed, N.S.; Abd-Allah, A.A.-R. Safety and outcome of treatment of metastatic melanoma using 3-bromopyruvate: A concise literature review and case study. Chin. J. Cancer. 2014, 33, 356–364.
- Rai, Y.; Yadav, P.; Kumari, N.; Kalra, N.; Bhatt, A.N. Hexokinase II inhibition by 3-bromopyruvate sensitizes myeloid leukemic cells K-562 to anti-leukemic drug, daunorubicin. Biosci. Rep. 2019, 39, 1–18.
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H.; Kunjithapatham, R.; Buijs, M.; Vossen, J.A.; Tchernyshyov, I.; Cole, R.N.; Syed, L.H.; Rao, P.P.; Ota, S.; et al. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is pyruvylated during 3-bromopyruvate mediated cancer cell death. Anticancer Res. 2009, 29, 4909–4918.
- Fan, T.; Sun, G.; Sun, X.; Zhao, L.; Zhong, R.; Peng, Y. Tumor energy metabolism and potential of 3-bromopyruvate as an inhibitor of aerobic glycolysis: Implications in tumor treatment. Cancers 2019, 11, 317.
- Attia, Y.M.; El-Abhar, H.S.; Al Marzabani, M.M.; Shouman, S.A. Targeting glycolysis by 3-bromopyruvate improves tamoxifen cytotoxicity of breast cancer cell lines. BMC Cancer 2015, 15, 838.
- Fischer, G.; Sieber, M.; Schellenberger, A. The carbonyl reactivity of 3-bromopyruvate and related compounds. Bioorg. Chem. 1982, 11, 478–484.
- Sadowska-Bartosz, I.; Szewczyk, R.; Jaremko, L.; Jaremko, M.; Bartosz, G. Anticancer agent 3-bromopyruvic acid forms a conjugate with glutathione. Pharmacol. Rep. 2016, 68, 502–505.
- Goede, V.; Eichhorst, B.; Fischer, K.; Wendtner, C.-M.; Hallek, M. Past, present and future role of chlorambucil in the treatment of chronic lymphocytic leukemia. Leuk. Lymphoma 2015, 56, 1585–1592.
- Leblond, V.; Johnson, S.; Chevret, S.; Copplestone, A.; Rule, S.; Tournilhac, O.; Seymour, J.F.; Patmore, R.D.; Wright, D.; Morel, P.; et al. Results of a randomized trial of chlorambucil versus fludarabine for patients with untreated Waldenström macroglobulinemia, marginal zone lymphoma, or lymphoplasmacytic lymphoma. J. Clin. Oncol. 2013, 31, 301–307.
- Ardeshna, K.M.; Smith, P.; Norton, A.; Hancock, B.W.; Hoskin, P.J.; MacLennan, K.A.; Marcus, R.E.; Jelliffe, A.; Vaughan Hudson, G.; Linch, D.C. Long-term effect of a watch and wait policy versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma:a randomised controlled trial. Lancet 2003, 362, 516–522.
- Parker, L.J.; Ciccone, S.; Italiano, L.C.; Primavera, A.; Oakley, A.J.; Morton, C.J.; Hancock, N.C.; Lo Bello, M.; Parker, M.W. The anti-cancer drug chlorambucil as a substrate for the human polymorphic enzyme glutathione transferase P1-1: Kinetic properties and crystallographic characterisation of allelic variants. J. Mol. Biol. 2008, 380, 131–144.
- Cobb, D.; Boehlert, C.; Lewis, D.; Armstrong, R.N. Stereoselectivity of isozyme C of glutathione S-transferase toward arene and azaarene oxides. Biochemistry 1983, 22, 805–812.
- Andrew, A.W.; Hayes, W.; Kruger, C.L. Hayes’ Principles and Methods of Toxicology; CRC Press, LLC: Boca Raton, FL, USA, 2014; p. 71.
- Hassan, F.; Preiss, R. Cyclophosphamide and related anticancer drugs. J. Chromatogr. B Biomed. Appl. 2001, 764, 173–192.
- Singh, R.K.; Kumar, S.; Prasad, D.N.; Bhardwaj, T.R. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401–433.
- Colvin, O.M. An overview of cyclophosphamide development and clinical applications. Curr. Pharm. Des. 1999, 5, 555–560.
- Hubbard, R.D.; Fidanze, S. 7.06—Alkylating and Platinum Antitumor Compounds. In Comprehensive Medicinal Chemistry II.; Taylor, J.B., Triggle, D.J., Eds.; Elsevier Ltd.: Oxford, UK, 2007; pp. 129–148.
- Ahlmann, M.; Hempel, G. The effect of cyclophosphamide on the immune system: Implications for clinical cancer therapy. Cancer Chemother. Pharmacol. 2016, 78, 661–671.
- Bagley, C.M.; Bostick, F.W.; DeVita, V.T. Clinical pharmacology of cyclophosphamide. Cancer Res. 1973, 33, 226–233.
- Li, F.; Patterson, A.D.; Höfer, C.C.; Krausz, K.W.; Gonzalez, F.J.; Idle, J.R. Comparative metabolism of cyclophosphamide and ifosfamide in the mouse using UPLC-ESI-QTOFMS-based metabolomics. Biochem. Pharmacol. 2010, 80, 1063–1074.
- Groehler, A.; Villalta, P.W.; Campbell, C.; Tretyakova, N. Covalent DNA-protein cross-linking by phosphoramide mustard and nornitrogen mustard in human cells. Chem. Res. Toxicol. 2016, 29, 190–202.
- Mills, K.A.; Chess-Williams, R.; McDermott, C. Novel insights into the mechanism of cyclophosphamide-induced bladder toxicity: Chloroacetaldehyde′s contribution to urothelial dysfunction in vitro. Arch. Toxicol. 2019, 93, 3291–3303.
- Yuan, Z.M.; Smith, P.B.; Brundrett, R.B.; Colvin, M.; Fenselau, C. Glutathione conjugation with phosphoramide mustard and cyclophosphamide. A mechanistic study using tandem mass spectrometry. Drug Metab. Dispos. 1991, 19, 625–629.
- Cohen, N.A.; Egorin, M.J.; Snyder, S.W.; Ashar, B.; Wietharn, B.E.; Pan, S.S.; Ross, D.D.; Hilton, J. Interaction of N,N′,N″-triethylenethiophosphoramide and N,N’,N″-triethylenephosphoramide with cellular DNA. Cancer Res. 1991, 51, 4360–4366.
- Van der Wall, E.; Beijnen, J.H.; Rodenhuis, S. High-dose chemotherapy regimens for solid tumors. Cancer Treat. Rev. 1995, 21, 105–132.
- Van Maanen, M.J.; Smeets, C.J.M.; Beijnen, J.H. Chemistry, pharmacology and pharmacokinetics of N,N′,N″-triethylenethiophosphoramide (ThioTEPA). Cancer Treat. Rev. 2000, 26, 257–268.
- Torabifard, H.; Fattahi, A. DFT study on Thiotepa and Tepa interactions with their DNA receptor. Struct. Chem. 2013, 24, 1–11.
- Jacobson, P.A.; Green, K.; Birnbaum, A.; Remmel, R.P. Cytochrome P450 isozymes 3A4 and 2B6 are involved in the in vitro human metabolism of thiotepa to TEPA. Cancer Chemother. Pharmacol. 2002, 49, 461–467.
- Dirven, H.A.A.M.; Dictus, E.L.J.T.; Broeders, N.L.H.L.; Van Ommen, B.; Van Bladeren, P.J. The role of human glutathione S-transferase isoenzymes in the formation of glutathione conjugates of the alkylating cytostatic drug thiotepa. Cancer Res. 1995, 55, 1701–1706.
- Schwöbel, J.A.H.; Wondrousch, D.; Koleva, Y.K.; Madden, J.C.; Cronin, M.T.D.; Schüürmann, G. Prediction of Michael-type acceptor reactivity toward glutathione. Chem. Res. Toxicol. 2010, 23, 1576–1585.
- Ruzza, P.; Calderan, A. Glutathione transferase (GST)-activated prodrugs. Pharmaceutics 2013, 5, 220–231.
- Schultz, T.W.; Yarbrough, J.W.; Hunter, R.S.; Aptula, A.O. Verification of the structural alerts for Michael acceptors. Chem. Res. Toxicol. 2007, 20, 1359–1363.
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, more than just another topoisomerase II poison. Med. Res. Rev. 2016, 36, 248–299.
- Hainswort, J.D.; Andrews, M.B.; Johnson, D.H.; Greco, F.A. Mitoxantrone, fluorouracil, and high-dose leucovorin: An effective, well-tolerated regimen for metastatic breast cancer. J. Clin. Oncol. 1991, 9, 1731–1735.
- Basch, E.M.; Scholz, M.; De Bono, J.S.; Vogelzang, N.; De Souza, P.; Marx, G.; Vaishampayan, U.; George, S.; Schwarz, J.K.; Antonarakis, E.S.; et al. Cabozantinib versus mitoxantrone-prednisone in symptomatic metastatic castration-resistant prostate cancer: A randomized Phase 3 trial with a primary pain endpoint. Eur. Urol. 2019, 75, 929–937.
- Nastoupil, L.J.; McLaughlin, P.; Feng, L.; Neelapu, S.S.; Samaniego, F.; Hagemeister, F.B.; Ayala, A.; Romaguera, J.E.; Goy, A.H.; Neal, E.; et al. High ten-year remission rates following rituximab, fludarabine, mitoxantrone and dexamethasone (R-FND) with interferon maintenance in indolent lymphoma: Results of a randomized study. Br. J. Haematol. 2017, 177, 263–270.
- Advani, A.S.; Cooper, B.; Visconte, V.; Elson, P.; Chan, R.; Carew, J.; Wei, W.; Mukherjee, S.; Gerds, A.; Carraway, H.; et al. A Phase I/II trial of MEC (Mitoxantrone, Etoposide, Cytarabine) in combination with ixazomib for relapsed refractory acute myeloid leukemia. Clin. Cancer. Res. 2019, 25, 4231–4237.
- Mewes, K.; Blanz, J.; Ehninger, G.; Gebhardt, R.; Zeller, K.-P. Cytochrome P-450-induced cytotoxicity of mitoxantrone by formation of electrophilic intermediates. Cancer Res. 1993, 53, 5135–5142.
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1--1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 27, 7066–7083.
- Komla-Ebri, D.; Dambroise, E.; Kramer, I.; Benoist-Lasselin, C.; Kaci, N.; Le Gall, C.; Martin, L.; Busca, P.; Barbault, F.; Graus-Porta, D.; et al. Tyrosine kinase inhibitor NVP-BGJ398 functionally improves FGFR3-related dwarfism in mouse model. J. Clin. Investig. 2016, 126, 1871–1884.
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282.
- Al-Shakliah, N.S.; Attwa, M.W.; Kadi, A.A.; AlRabiah, H. Identification and characterization of in silico, in vivo, in vitro, and reactive metabolites of infigratinib using LC-ITMS: Bioactivation pathway elucidation and in silico toxicity studies of its metabolites. RSC Adv. 2020, 10, 16231–16244.
- Zhao, Z.; Koeplinger, K.A.; Padbury, G.E.; Hauer, M.J.; Bundy, G.L.; Banitt, L.S.; Schwartz, T.M.; Zimmermann, D.C.; Harbach, P.R.; Mayo, J.K. Bioactivation of 6,7-dimethyl-2,4-di-1-pyrrolidinyl-7h-pyrrolopyrimidine (U-89843) to reactive intermediates that bind covalently to macromolecules and produce genotoxicity. Chem. Res. Toxicol. 1996, 9, 1230–1239.
- Lyss, G.; Schmidt, T.J.; Merfort, I.; Pahl, H.L. Helenalin, an anti-inflammatory sesquiterpene lactone from Arnica, selectively inhibits transcription factor NF-κB. Biol. Chem. 1997, 378, 951–962.
- Lim, C.B.; Fu, P.Y.; Ky, N.; Zhu, H.S.; Feng, X.L.; Li, J.; Srinivasan, K.G.; Hamza, M.S.; Zhao, Y. NF-κB p65 repression by the sesquiterpene lactone, helenalin, contributes to the induction of autophagy cell death. BMC Complement. Altern. Med. 2012, 12, 93.
- Jakobs, A.; Steinmann, S.; Henrich, S.M.; Schmidt, T.J.; Klempnaue, K.H. Helenalin acetate, a natural sesquiterpene lactone with anti-inflammatory and anti-cancer activity, disrupts the cooperation of CCAAT box/enhancer-binding protein β (C/EBPβ) and co-activator p300. J. Biol. Chem. 2016, 9, 26098–26108.
- Jürgens, F.M.; Behrens, M.; Humpf, H.-U.; Robledo, S.M.; Schmidt, T.J. In vitro metabolism of helenalin acetate and 11α,13-dihydrohelenalin acetate: Natural sesquiterpene lactones from Arnica. Metabolites 2022, 12, 88.
- Schmidt, T.J. Glutathione adducts of helenalin and 11α,13-dihydrohelenalin acetate inhibit glutathione S-transferase from horse liver. Planta Med. 2000, 66, 106–109.
- Lang, W.; Caldwell, G.W.; Masucci, J.A. Evaluation of the effect of oxygen exposure on human liver microsomal metabolism of mitomycin C in the presence of glutathioneusing liquid chromatography–quadrupole time of flight mass spectrometry. Anal. Biochem. 2005, 343, 268–276.
- Verweij, J.; Pinedo, H.M. Mitomycin C: Mechanism of action, usefulness and limitations. Anticancer Drugs 1990, 1, 5–13.
- Tomasz, M. Mitomycin C: Small, fast and deadly (but very selective). Chem. Biol. 1995, 2, 575–579.
- Avendaño, C.; Menéndez, J.C. Chapter 6Anticancer Drugs That Interact with the DNA Minor Groove. In Medicinal Chemistry of Anticancer Drugs, 2nd ed.; Avendaño, C., Menéndez, J.C., Eds.; Elsevier Ltd.: Oxford, UK, 2015; pp. 243–271.
- Sharma, M.; Tomasz, M. Conjugation of glutathione and other thiols with bioreductively activated mitomycin C. Effect of thiols on the reductive activation. Chem. Res. Toxicol. 1994, 7, 390–400.
- Shibata, Y.; Chiba, M. The role of extrahepatic metabolism in the pharmacokinetics of the targeted covalent inhibitors afatinib, ibrutinib, and neratinib. Drug Metab. Dispos. 2015, 43, 375–384.
- Rood, J.J.M.; Dormans, P.J.A.; Van Haren, M.J.; Schellens, J.H.M.; Beijnen, J.H.; Sparidans, R.W. Bioanalysis of ibrutinib, and its dihydrodiol- and glutathione cycle metabolites by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2018, 1090, 14–21.
- Piesche, M.; Roos, J.; Kühn, B.; Fettel, J.; Hellmuth, N.; Brat, C.; Maucher, I.V.; Awad, O.; Matrone, C.; Comerma Steffensen, S.G.; et al. The emerging therapeutic potential of nitro fatty acids and other Michael acceptor-containing drugs for the treatment of inflammation and cancer. Front. Pharmacol. 2020, 11, 1297.