Glutathione is the predominant intracellular thiol-containing tripeptide found in all animal tissues, plants, fungi, and some microorganisms [
27,
28]. Under physiological conditions, it is mainly present in the cytoplasm in the reduced form (GSH), which is also the biologically active form. GSH is less easily oxidized than its precursors, cysteine and γ-glutamylcysteine; the fully oxidized form with a disulfide between two identical GSH molecules (GSSG) represents less than 1% of the total GSH pool in the cell [
29]. GSH concentration in human cells typically ranges from 0.1 to 10 mmol/L, being most focused in the liver (up to 10 mmol/L), spleen, kidney, lens, erythrocytes, and leukocytes [
30,
31], wherein its depletion and/or altered level are associated with various diseases, including cancer, cardiovascular, inflammatory, immune, metabolic, and neurodegenerative diseases [
32]. Maintaining optimal GSH:GSSG ratios in the cell is critical to survival; hence, tight regulation of this system is imperative [
33].
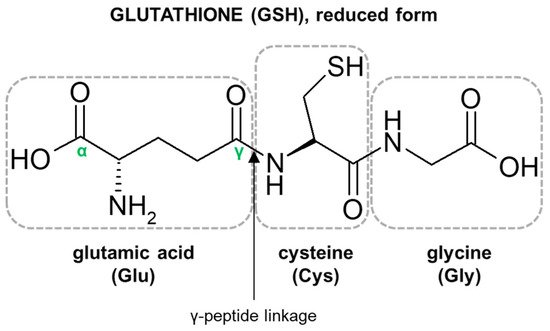
The characteristic structural features of the GSH molecule (
Figure 1) determine its many diverse biological functions. In contrast to an α-peptide linkage usually found in a number of endogenous peptides, the L-glutamic acid (Glu) and L-cysteine (Cys) of GSH are joined by an unusual γ-peptide linkage [
28]. Such a bond promotes GSH resistance to hydrolysis by most intracellular aminopeptidases as well as provides for specificity in other GSH-enzyme interactions [
34]. In turn, the activity of the high electron-donating sulfhydryl (thiol, -SH) group of Cys residue supports the reducing properties of GSH by way of a thiol-exchange system (-SH to -S-S-), enabling the participation of GSH in intracellular antioxidative and detoxifying reactions [
35]. The reactivity of -SH is due to the thiolate anion (S
−), the relative concentration of which is regulated by the acidity of thiol (pK
a = 9.2). At physiological pH, for every 100 GSH molecules in the -SH state, approximately 3.7 are in a thiolate form [
36]. Due to the polarizability of the sulfur atom, GSH is a strong ‘soft’ nucleophile, and unlike other phase II enzyme cofactors such as uridine 5′-diphosphoglucuronic acid (UDPGA) and 3′-phosphoadenosine-5′-phosphosulfate (PAPS), it easily reacts with various ‘soft’ electrophiles [
10], which may also be anticancer drugs. Net-negative charge of cysteinyl residue and overall GSH hydrophilicity greatly increase the aqueous solubility of the lipophilic moieties with which it becomes conjugated. GSH S-conjugates usually achieve a molecular weight higher than 300–500 g/mol (average molecular weight of GSH = 307.3235 g/mol) and are thus preferentially secreted via the biliary system. Then, the final cysteinyl conjugates are reabsorbed into the liver, from where they travel to the kidney for acetylation and excretion as a mercapturic acid [
37].
Reduced GSH has been adopted through evolution to perform multiple significant cellular functions in living organisms. It is responsible for the correct thiol–disulfide balance and the associated oxidation-reduction potential of cells [
38]. The biologically important role of GSH is related to the possibility of regeneration of the -SH moieties of proteins, which counteracts the effects of oxidative reactions, inactivating cell proteins [
39]. This compound is also involved in the reduction of ribonucleotides to deoxyribonucleotides, i.e., it has a direct impact on DNA biosynthesis and the related proliferation process [
8]. Moreover, it mediates in the synthesis of proteins and in amino acid transport [
40]. Further, GSH serves as a reservoir and transporter of cysteine [
41], a regulator of calcium ion homeostasis [
42], a versatile cofactor for many cytoplasmic enzymes [
8], and it is a link in the mitochondrial mechanism to cell death [
43].
In addition to the functions mentioned, GSH is an important component of the system that detoxifies both electrophilic xenobiotics and metabolically produced free radicals, i.e., reactive oxygen species (ROS), by the formation of GSH S-conjugates. Thus, it plays a central role in the protection of cells against a variety of exogenous and endogenous potentially harmful compounds [
28,
44]. The reactions to form GSH S-conjugates may be non-enzymatic, although they are greatly accelerated by GSH-dependent enzymes such as GSTs [
11]. Hence, the effectiveness of the detoxification pathway depends upon the intracellular concentration of GSH, the presence of GSTs of appropriate specificity, and/or the capacity of the cell for rapid resynthesis of GSH [
45]. Conjugation reactions of GSH with electrophilic compounds to GSH S-conjugates occur mainly in the liver, which exports GSH and has the highest GST activity [
28]. Although by conjugation with GSH many compounds are rendered less toxic than the original parent xenobiotic, it has also been reported that some drugs, including those with anticancer activity, become more reactive following this reaction. Thus, GSH conjugation may also play an important role in drug bioactivation processes [
18,
19,
20].
3. Overview of Mechanisms of Glutathione Conjugation of Anticancer Drugs
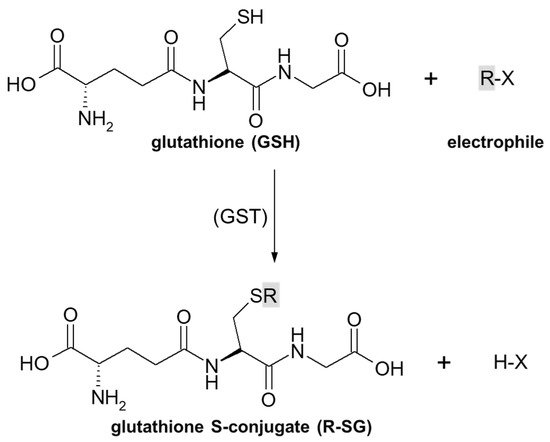
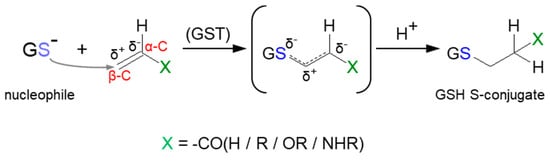
As previously stated, the important part of the GSH molecule in terms of its chemical activity is the nucleophilic sulfhydryl (thiol, -SH) group of the cysteine residue. Generally, as illustrated in
Figure 4, conjugation with GSH involves attachment of the xenobiotic molecule or its phase I metabolite (assigned as R-X) with this group to form the GSH S-conjugate (assigned as R-SG). In fact, the reactive nucleophilic species is the thiolate anion of GSH (GS
−), whose concentrations at physiological pH are approximately 1% of GSH concentration (pK
a of GSH = 9.2) [
38]. Due to the low electronegativity and high polarizability of the sulfur atom, GSH acts as a ‘soft’ nucleophile and, as such, can be used to sense the reactivity of ‘soft’ electrophiles (i.e., compounds that contain an acceptor atom with high polarizability, low electronegativity, and often unshared
p or
d valence electrons) [
51]. Xenobiotics that are conjugated with GSH are either very electrophilic right away or are metabolized to electrophilic compounds. Some reactions of the tripeptide GSH with cellular electrophiles have spontaneous rates which vary considerably depending on the reactivity of the electrophile, and frequently, but not always, are catalyzed by various GST isoenzymes [
34]. However, non-enzymatic reactions are usually much slower than those catalyzed by GSTs.
Figure 4. General scheme of glutathione (GSH) conjugation to a generic electrophile (R-X) to form the glutathione S-conjugate (R-SG). The reaction may be catalyzed by the enzyme glutathione S-transferase (GST).
3.1. Nucleophilic Substitution
Substitution reactions, which involve the reaction of a nucleophile with an electrophile, are prevalent in physiological and metabolic processes, in the action of some drugs, and in the chemical synthesis of nearly all drugs [
57]. Thus, nucleophilic substitution is also the basic and the most widespread mechanism of GSH conjugation of electrophilic compounds. It is observed in several families of anticancer drugs, including alkyl, allylic, benzylic, and aryl halides, nitrogen mustard derivatives, or platinum complexes [
33]. Electrophiles are positively charged or have a polarized bond with a partial positive character. Electrophiles capable of undergoing substitution reactions have a leaving group—a species that can accept and stabilize the pair of electrons that make up the bond being broken [
57].
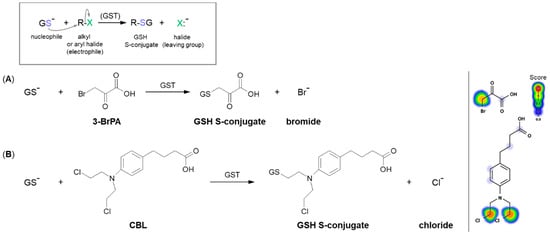
A common type of nucleophilic substitution reaction is the bimolecular nucleophilic substitution reaction, or S
N2 reaction for short, where one atom or functional group is replaced with an electronegative GSH molecule (the frame in
Figure 5). In this case, bond formation and bond breaking occur simultaneously and the leaving group tends to be a weaker base than the nucleophile. Halide ions, such as I
−, Br
−, and Cl
−, are very good leaving groups and thus give fast reactions [
58].
Figure 5. Reaction schemes of nucleophilic substitution of the thiolate anion of GSH (GS
–) to the halogen atom (leaving group) in (
A) 3-bromopyruvic acid (3-BrPA) [
17] and (
B) chlorambucil (CBL) [
65]. The frame shows the general scheme of the GSH conjugation reaction. Predicted bioactive sites of GSH conjugation for anticancer drugs were obtained by XenoSite Reactivity Predictor available at
https://swami.wustl.edu/xenosite/p/reactivity (accessed on 18 May 2022).
3.1.1. Halogen Atom as a Leaving Group
The S
N2 reaction mechanism of alkyl halide with GSH can be illustrated by the conjugation of 3-bromopyruvic acid (3-BrPA) with GSH (
Figure 5A). 3-BrPA is the brominated derivative of pyruvic acid with proven anticancer activity against many different cancers in children and adults [
59,
60]. Like other α-bromoketones, it is widely known as a strong alkylating agent with a high affinity for protein binding as well as an antimetabolite [
17]. Moreover, being an inhibitor of key glycolysis enzymes, including hexokinase II [
61] and glyceraldehyde 3-phosphate dehydrogenase [
62], 3-BrPA inhibits the growth of neoplastic cells that perform aerobic glycolysis known as the ‘Warburg effect’ [
63]. Additionally, it also selectively blocks mitochondrial oxidative phosphorylation, angiogenesis, and energy production in cancer cells [
63,
64].
Based on a chemical view, the thiolate anion of GSH (GS
−) easily attacks the carbon atom to which the bromine atom is attached. This reaction occurs because of the imbalance of the electron density between the carbon and halogen since it is a polar covalent bond. The more electronegative bromine atom pulls the electron density, thus making the carbon partially positively charged (an electrophilic center) and susceptible to a nucleophilic GS
− attack [
66]. A bromine atom built into the structure of the 3-BrPA molecule is a good leaving group because the negatively charged bromine atom (bromide) is stable enough to exist on its own when it leaves the molecule. Hence, the conjugation to GSH does not require any prior metabolic activation of the parent compound. 3-BrPA was reported to form GSH S-conjugate both under GST catalysis and also in an enzyme-free system. Further, it is eliminated through the mercapturic acid synthesis pathway where it is excreted from the cell by ATP-binding cassette transporter proteins [
17,
67].
Another example of a similar S
N2 reaction is the GSH conjugation of chlorambucil (CBL; the brand name: Leukeran) (
Figure 5B). CBL is an alkylating agent approved for use in various malignant and non-malignant neoplasms, such as chronic lymphocytic leukemia [
68], lymphosarcoma [
69], and giant follicular lymphoma [
70]. In the presence of GSH, CBL behaves as an efficient substrate for GSTA1-1 and GSTP1-1 isoenzymes [
65,
71]. As with the 3-BrPA, the formation of the corresponding GSH S-conjugate undergoes without prior CBL activation as the negatively charged chlorine atom (chloride) is a good enough leaving group. Moreover, kinetic data suggest that the rate-limiting of the catalytic reaction between CBL and GSH is the reaction product release [
65].
3.1.2. Tensioned Ring-Opening Reaction
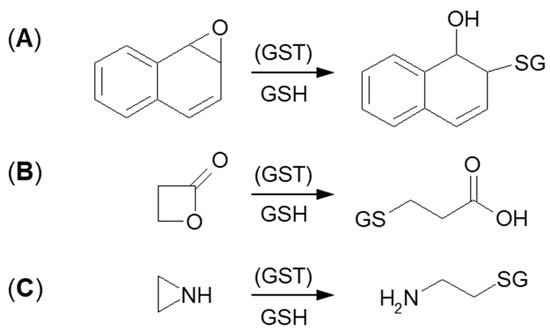
The next type of GSH conjugation mechanism is the attachment of the thiolate anion (GS
–) to the epoxide, four-membered lactone, or three-membered aziridine resulting in ring-opening (
Figure 6). Such reactions take place quite easily because the rings are composed of three or four carbon atoms which are highly tensed and their energy is quite high. Additionally, such a process will be easier, if the tensed system includes a heteroatom, because it will inductively decrease the electron density on adjacent carbon atoms [
72,
73]. Under basic conditions, ring-opening occurs by an S
N2 mechanism, and the less substituted carbon is the site of GS
– nucleophilic attack. The described transformation can take place stereoselectively, depending on the GST isoenzyme that catalyzes a reaction.
Figure 6. GSH conjugation via opening the tensioned ring of (A) epoxy, (B) lactone, and (C) aziridine.
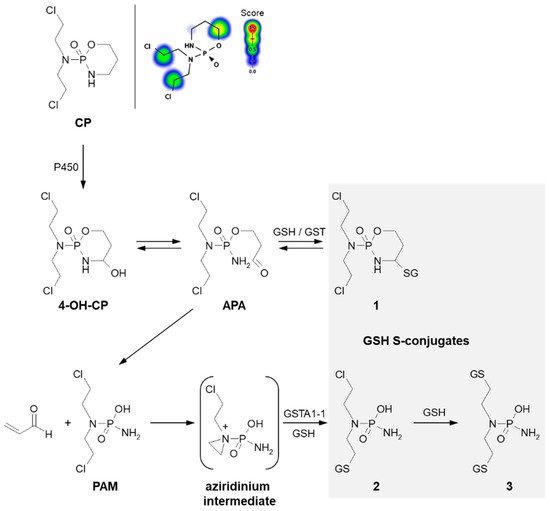
Formation of GSH S-conjugate, accompanied by the aziridine ring-opening, takes place, e.g., in the case of cyclophosphamide and thiotepa. Cyclophosphamide (CP; the brand name: Cytoxan) belongs to the alkylating agent and nitrogen mustard family of medications [
74,
75]. Its mechanism of action, quite similar to that of chlorambucil, relies on interfering with the duplication of DNA and RNA synthesis [
76]. It is a chemotherapeutic approved to treat malignant lymphomas, neuroblastoma, multiple myeloma, leukemias, ovarian, breast, and many other cancers [
77]. Such a wide spectrum of clinical uses makes it an essential component of numerous combination chemotherapeutic regimens. Moreover, CP is also used to suppress the immune system [
78].
A prerequisite for conjugation with GSH is the presence of a reactive electrophilic metabolite(s). Unlike 3-BrPA and CBL, CP must undergo previous metabolic activation by hepatic microsomal mixed-function oxidases (i.e., P450s, mainly 2B6, 2C8, and 2C9) (
Figure 7) [
79,
80]. The generally accepted mechanism for the generation of active metabolites of CP involves oxidation to the primary 4-hydroxycyclophosphamide (4-OH-CP) metabolite, which stays in tautomeric equilibrium with the ring-opened aldophosphamide (APA). Then, non-enzymatic cleavage of APA results in the formation of two toxic species—phosphoramide mustard (PAM) and acrolein. PAM is believed to unfold cell toxicity by DNA alkylation [
81] while acrolein is held to be responsible for some aspects of host toxicity, such as hemorrhagic cystitis [
82]. Therefore, GSH conjugation can result in the formation of three types of GSH S-conjugates, i.e., 4-monoglutathionyl CP (4-GSCP) (
1), mono- (
2) and diglutathionyl PAMs (
3) [
13,
83]. The formation of 4-GSCP was found to be reversible, and by APA hydrolysis, PAM was formed. Thus, 4-GSCP can be considered a stable reservoir for the generation of PAM that subsequently undergoes two consecutive GSH conjugations. The reaction with nucleophilic GSH was established to proceed through the positively charged and highly polarized aziridinium ion (aziridinium intermediate) that can be opened in both the enzyme-catalyzed and the chemical reactions. On the other hand, the second GSH conjugation reaction was shown to occur through the direct displacement of chloride [
13]. The 4-GSCP formation reaction can be catalyzed by various GSTs isoenzymes, whereas GST1A-1 has the greatest effect on the rate of monoglutathionyl PAM formation. Melphalan, mechlorethamine, ifosfamide, carmustine, lomustine, and nimustine are examples of other anticancer-active nitrogen mustard derivatives that react with GSH in a manner similar to that described for CP [
13,
80].
Figure 7. Scheme of cyclophosphamide (CP) activation and drug reaction with GSH [
74]. Possible GSH S-conjugates:
1: 4-monoglutathionyl cyclophosphamide,
2: monoglutathionyl phosphoramide mustard,
3: diglutathionyl phosphoramide mustard. 4-OH-CP = 4-hydroxycyclophosphamide; APA = aldophosphamide; PAM = phosphoramide mustard; P450 = cytochrome P450. Predicted bioactive sites of GSH conjugation for anticancer drug were obtained by XenoSite Reactivity Predictor available at
https://swami.wustl.edu/xenosite/p/reactivity (accessed on 18 May 2022).
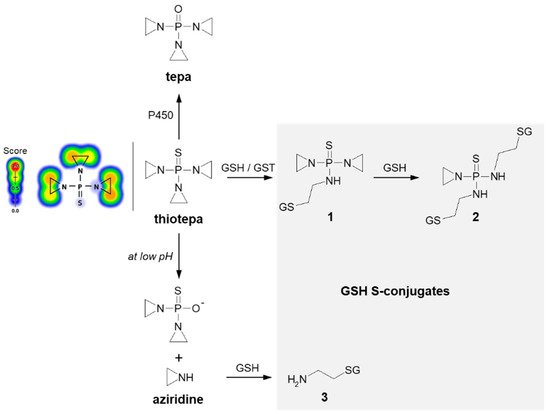
Thiotepa (N,N′,N″-triethylenethiophosphoramide; the brand name: Tepadina) also acts as an alkylating agent [
84]. Being active against a wide variety of cancers, it is commonly used in combination with other chemotherapeutic agents to treat ovarian cancer, bladder cancer, and breast cancer [
85,
86]. Chemically, it is an organophosphorus compound containing a four-coordinated phosphorus atom and three aziridine moieties (
Figure 8) through which the drug probably induces crosslinks with DNA, interfering with DNA replication and cell division [
87]. The major metabolite formed from thiotepa during P450-catalyzed transformation (i.e., desulfuration reaction) is N,N′,N″-triethylenephosphoramide (tepa) [
88]. In turn, when reacting with GSH, the tensioned ring of thiotepa containing a nitrogen atom as a heteroatom is opened, leading to the formation of mono- (
1) and diglutathionyl thiotepa (
2). In the metabolic pathway of thiotepa, 2-aminoethyl GSH (
3), which is the product of the direct GSH conjugation of aziridine, was also characterized. The results confirmed that only thiotepa but not its monoglutathionyl S-conjugate is a substrate for GSTs (mainly A1-1 and P1-1 isoenzymes). Moreover, the non-enzymatic reaction of the aziridinium moieties of thiotepa with GSH is strongly dependent on the pH, and the yield of the reaction is greatest at low pH [
89].
Figure 8. Scheme of thiotepa activation and drug reaction with GSH [
89]. Possible GSH S-conjugates:
1: monoglutathionyl thiotepa,
2: diglutathionyl thiotepa,
3: 2-aminoethyl glutathione. P450 = cytochrome P450. Predicted bioactive sites of GSH conjugation for anticancer drug were obtained by XenoSite Reactivity Predictor available at
https://swami.wustl.edu/xenosite/p/reactivity (accessed on 18 May 2022).
3.2. Nucleophilic Addition (Michael Addition)
Some reactions leading to the formation of GSH S-conjugates occur through Michael addition (or conjugate addition). Such a mechanism applies to α,β-unsaturated compounds (the so-called Michael acceptors) characterized by having carbon–carbon double (C=C) or carbon–carbon triple (C≡C) bonds with a strongly electron-withdrawing substituent(s) (e.g., a carbonyl, carboxyl, or nitro group) (
Figure 12) [
114]. This results in a polarizable electron density at the π bond, where the β-carbon atom (β-C) is positively polarized and becomes the preferred site of an attack of a soft nucleophile (Michael donor), e.g., the thiol group of cysteine in GSH. Although many of them can form thioethers non-enzymatically, GST-catalyzed Michael addition is much faster [
10,
115]. Compounds possessing Michael acceptor units feature a broad spectrum of bioactivity. They are considered to be particularly reactive and are thus capable of bonding with biological macromolecules [
116].
Figure 12. General scheme of Michael addition (or conjugate addition) to α,β-unsaturated compound with carbon−carbon double (C=C) bond.
Mitoxantrone (MTX; the brand name: Novantrone), a synthetic anthraquinone antineoplastic agent, is an example of a compound that undergoes GSH conjugation by Michael addition. MTX is a potent type II topoisomerase inhibitor that disrupts DNA synthesis and DNA repair in both healthy cells and cancer cells by intercalation between DNA bases [
117]. It is commonly applied in the treatment of breast [
118] and prostate [
119] cancers, lymphomas [
120], and leukemias, primarily acute myeloid leukemia [
121], with excellent efficacy.
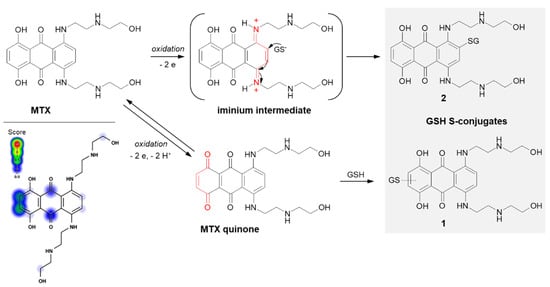
In the case of MTX, at least two GSH conjugation pathways were identified (
Figure 13). MTX is known to resist reductive enzymatic activation but is subject to facile oxidative enzymatic action [
122]. The development of GSH-dependent resistance provides further evidence that the oxidative activation may be a relevant mode of drug action. The in vivo and in vitro studies of Mewes et al. [
122] performed using minipigs, cultured rat hepatocytes, and human HepG2 hepatoma cells, respectively, revealed the formation of a major monoglutathionyl MTX (
1) and its various degradation products. The ability of MTX to react with GSH enables the formation of an MTX quinone derivative by two-electron oxidation of the parent drug, which is in the form of a hydroquinone. Additionally, there are also data confirming the possibility of the formation of another MTX-GSH S-conjugate (
2) [
117]. Following the MTX enzymatic oxidation process within the aromatic ring containing polyamine side chains, the intramolecular Michael addition occurs. Presumably, the mechanism of this GSH conjugation reaction takes place via a labile iminium ion intermediate which activates the aromatic ring towards the attack of the external cellular nucleophile (e.g., GSH, DNA).
Figure 13. Scheme of mitoxantrone (MTX) activation and drug reaction with GSH [
16,
117]. The Michael acceptor moieties are marked in red. Predicted bioactive sites of GSH conjugation for anticancer drug were obtained by XenoSite Reactivity Predictor available at
https://swami.wustl.edu/xenosite/p/reactivity (accessed on 18 May 2022).
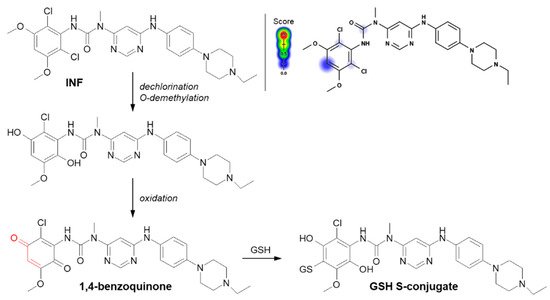
Another example of a Michael addition-type reaction is infigratinib (INF, NVP-BGJ398). It is a novel small-molecule chemotherapeutic drug used for first-line treatment of advanced or metastatic cholangiocarcinoma (bile duct cancer). It was discovered that INF inhibits human fibroblast growth factor receptors (FGFRs), which are a family of receptor tyrosine kinases that may be upregulated in different cancer cell types [
123]. For this reason, it is an investigational drug under development for the treatment of patients with various FGFR-driven diseases [
124,
125]. Al-Shakliah et al. [
126] detected at least three GSH S-conjugate metabolites of INF using liquid chromatography ion trap mass spectrometry (LC-ITMS). As shown in
Figure 14, the halogenated benzene ring of the INF structure undergoes metabolic bioactivation sequentially by dechlorination, O-demethylation, and oxidation to form the reactive 1,4-benzoquinone intermediate that is attacked by GSH [
126,
127].
Figure 14. Reaction scheme of infigratinib (INF) conjugation with GSH [
126]. A Michael acceptor moiety is marked in red. Predicted bioactive sites of GSH conjugation for anticancer drug were obtained by XenoSite Reactivity Predictor available at
https://swami.wustl.edu/xenosite/p/reactivity (accessed on 18 May 2022).
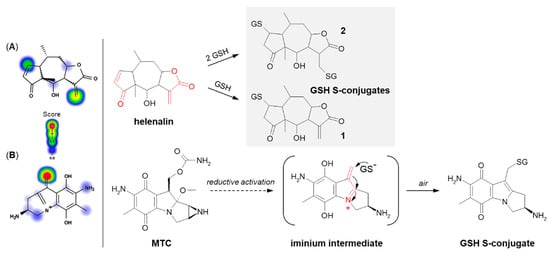
The next example is helenalin, a natural sesquiterpene lactone present in a large number of species mostly from the
Asteraceae family, which has a variety of observed effects in vitro, including anti-inflammatory and anticancer activities [
128,
129,
130]. As the compound structure contains reactive Michael acceptor systems (i.e., α,β-unsaturated ketone moiety and α,β-unsaturated lactone moiety), it is able to easily react with GSH [
131]. The 2β-mono- (
1) and 2,13β-diglutathionyl (
2) conjugates of helenalin were shown to be formed by spontaneous Michael addition at physiological pH (
Figure 15A). Interestingly, these were found to inhibit GST from horse liver, while free helenalin showed no inhibitory activity [
132].
Figure 15. Reaction schemes of (
A) helenalin [
131] and (
B) mitomycin C (MTC) [
133] conjugation with GSH. The Michael acceptor moieties are marked in red. Predicted bioactive sites of GSH conjugation for anticancer drugs were obtained by XenoSite Reactivity Predictor available at
https://swami.wustl.edu/xenosite/p/reactivity (accessed on 18 May 2022).
Mitomycin C (MTC) is a natural cytostatic antibiotic used as a chemotherapeutic agent by virtue of its anticancer activity. The mechanism of drug action is typical for that based on DNA alkylation [
134,
135]. Importantly, MTC requires previous activation via enzymatic reduction (bioreductive activation) to exert its biological effects. One-electron reduction steps to the corresponding semiquinone and then to hydroquinone initiates a cascade of consecutive reactions (i.e., spontaneous elimination of methanol from hydroquinone, elimination of the carbamate group, opening of the aziridine ring) that gives an unstable iminium intermediate which reacts with GSH through a Michael-type reaction [
133,
136] (
Figure 15B). MTC was shown to form both mono- and diglutathionyl conjugates. It was also found that GSH itself did not reduce MTC, and unreduced drug did not form conjugates with GSH [
137].
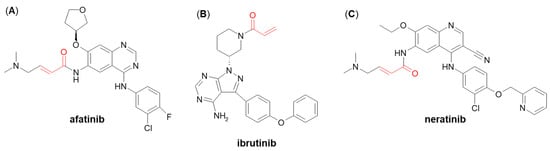
A few more examples of Michael acceptor-containing anticancer therapeutics metabolizing through GSH conjugation are afatinib, ibrutinib, and neratinib (
Figure 16) [
138,
139]. They are all inhibitors of various tyrosine kinases and use their own Michael acceptor moiety for irreversible binding to a free cysteine residue of the targeted protein. In recent years, such a targeted covalent modification of regulatory proteins by Michael acceptors became recognized as a promising approach to drug discovery [
140]. It can be expected that GSH plays an integral role in the clearance of these electrophilic drugs. Shibata and Chiba [
138] showed that both afatinib and neratinib undergo extensive conjugation with GSH in buffer and cytosolic subfraction deriving from liver and kidney tissues, whereas ibrutinib has exhibited much lower degree of GSH/GST-dependent conjugation [
138]. These findings may be helpful in optimizing pharmacokinetics in humans during the development stage of other targeted covalent inhibitors.
Figure 16. Chemical structures of (A) afatinib, (B) ibrutinib, and (C) neratinib. The Michael acceptor moieties are marked in red.