2. Osteopontin in Heart Failure
Embryogenesis is associated with the augmented expression of osteopontin in various tissues and organs, including the heart [
56,
57,
58]. Despite this implied function during embryogenesis, mice lacking osteopontin grow to maturity without any overt signs of morphological and functional cardiac abnormalities [
29,
59]. In line with this, in homeostatic conditions, osteopontin expression in the adult heart is very low [
58,
60,
61,
62,
63], although it is regulated in a cell-specific manner. For example, a low basal expression of osteopontin was reported in cardiomyocytes [
64], while it can be readily detected in coronary artery endothelial cells and cardiac fibroblasts [
64,
65]. Nevertheless, osteopontin-null mice respond to a variety of pathological stimuli, including tissue injury, inflammation, and infection, differently from wild-type mice [
66].
In the last two decades, several studies utilizing osteopontin depletion either by neutralizing antibodies or by targeted mutagenesis have further advanced our understanding of the role of osteopontin in various cardiac pathologies [
59,
67,
68,
69,
70,
71,
72]. Osteopontin-null mice were used in a number of HF models, including angiotensin-II (Ang-II) infusion [
59], aldosterone infusion [
67], transverse aortic constriction (TAC) [
68], desmin-deficient model of dilated cardiomyopathy (DCM) [
69], streptozotocin-induced model of diabetic cardiomyopathy [
70], left anterior descending artery (LAD) ligation as a model of myocardial infarction (MI) [
71] and a brief, repetitive LAD-occlusion model of ischemia-reperfusion (IR)-induced myocardial injury [
72]. However, these and other studies have yielded conflicting results suggesting disease- and cell-specific roles of osteopontin in cardiac pathologies.
2.1. Cell-Specific Regulation of Osteopontin
2.1.1. Osteopontin in Cardiomyocytes
Several factors have been shown to induce osteopontin expression in cardiomyocytes including aldosterone [
73], dexamethasone [
64], endothelin-1 [
58], norepinephrine (NE) [
58], and ROS [
74] (
Figure 2). In contrast, other known stimulators of osteopontin expression such as Ang-II [
65,
75], interleukin-1β (IL-1β), and interferon-γ (IFN-γ) [
64] failed to induce osteopontin expression in cardiomyocytes.
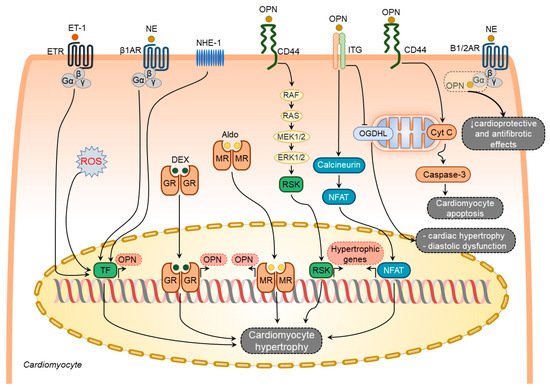
Figure 2. Osteopontin signaling in cardiomyocytes. Various factors such as endothelin-1 (ET-1), norepinephrine (NE), sodium-hydrogen exchanger isoform-1 (NHE-1), reactive oxygen species (ROS), dexamethasone (DEX), and aldosterone (Aldo) upregulate osteopontin (OPN) in cardiomyocytes. In turn, osteopontin regulates several processes in cardiomyocytes via modulating certain signaling pathways. These pathways include induction of hypertrophic genes expression via integrin- and CD44-mediated signaling pathways, cardiomyocyte hypertrophy and diastolic dysfunction via blocking mitochondrial protein oxoglutarate dehydrogenase-like (OGDHL), and cardiomyocyte apoptosis via mitochondrial apoptosis signaling pathway, attenuation of the beneficial effects of adrenergic signaling by directly binding and blocking G-α subunit of the adrenergic receptors. The overall effect of osteopontin signaling in cardiomyocytes results in cardiomyocyte hypertrophy, cardiomyocyte apoptosis, and cardiomyocyte mitochondrial dysfunction.
Endothelin-1 and NE-mediated osteopontin upregulation in rat cardiomyocytes coincide with the induction of atrial natriuretic peptide expression [
58], suggesting an active involvement of osteopontin in hypertrophic responses of the cardiomyocytes. Cardiomyocyte hypertrophy, LV dilatation, and dysfunction caused by Na
+/H
+ exchanger isoform 1 (NHE1) overexpression in mice were associated with osteopontin upregulation and osteopontin-CD44 mediated activation of p90-ribosomal S6 kinase signaling pathways [
76]. All these pathological changes were reversed in double transgenic mice expressing active NHE1 and osteopontin knockout [
76]. Hypertrophic effects of osteopontin on cardiomyocytes can also be mediated through the calcineurin/NFAT pathway [
77,
78]. In addition, osteopontin can diminish the cardioprotective and antifibrotic effects of β1- and β2-adrenoceptors signaling pathways by direct binding to their Gαs subunits [
73].
Several studies have demonstrated an association of increased osteopontin expression with enhanced cardiomyocyte apoptosis in different HF models [
29,
67,
70]. Cardiomyocyte-specific osteopontin overexpression in mice was associated with decreased LV function and increased cardiomyocyte apoptosis [
79]. In support of these findings, treatment of adult rat cardiomyocytes with purified osteopontin or adenoviral-mediated expression of osteopontin induced cell apoptosis via CD44-mediated activation of the mitochondrial death pathway and endoplasmic reticulum stress [
79].
Col4a3-deficiency in mice recapitulates multiple features of HF with preserved ejection fraction and is associated with cardiac hypertrophy, diastolic dysfunction, myocardial fibrosis, and mitochondrial dysfunction [
80]. Targeting osteopontin significantly ameliorated the HF phenotype in these mice [
80]. This study revealed that osteopontin induced mitochondrial dysfunction in cardiomyocytes by downregulating myocardial 2-oxoglutarate dehydrogenase-like, a vital protein in normal mitochondrial function [
80].
When taken together, osteopontin serves as an upstream and downstream of diverse signaling pathways involved in cardiomyocyte hypertrophy, apoptosis, and mitochondrial dysfunction (Figure 2).
2.1.2. Osteopontin in Cardiac Fibroblasts
In DCM patients, increased levels of myocardial osteopontin strongly correlated with collagen expression [
81], suggesting osteopontin involvement in cardiac fibrogenesis. Plasma from aged osteopontin-null mice failed to cause age-specific activation of cardiac fibroblasts [
82]. Similarly, cardiac fibroblasts isolated from osteopontin-null mice displayed decreased proliferation and adhesion to ECM [
59,
83], less spreading, less resistance to detachment by shear stress, and a reduction in collagen gel contraction, which could be partially restored by ectopic osteopontin expression [
83].
Several growth factors and cytokines have been shown to regulate osteopontin expression in cardiac fibroblasts, including Ang-II [
65,
75] and syndecan-4 [
84] (
Figure 3). Ang-II enhances osteopontin expression in cardiac fibroblasts via NADPH-ROS-mediated activation of ERK1/2 and JNKs pathways [
65]. Syndecan-4 increases osteopontin expression via calcineurin/NFAT signaling pathways [
84]. Mechanical stretch can also induce osteopontin expression in cardiac fibroblasts [
85].
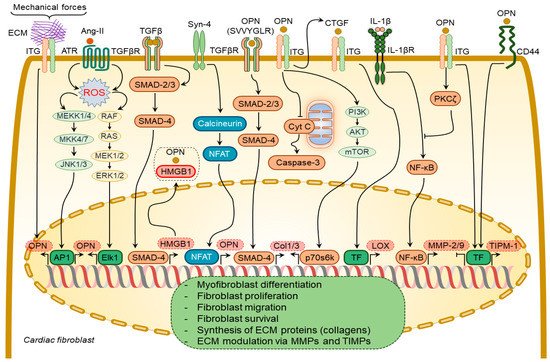
Figure 3. Osteopontin signaling in cardiac fibroblasts. A number of factors induce osteopontin (OPN) expression in cardiac fibroblasts including angiotensin-II (Ang-II), syndecan-4 (Syn-4), and mechanical forces. In turn, osteopontin regulates several processes in cardiac fibroblasts via modulating certain signaling pathways. These processes include binding of osteopontin to high mobility group box 1 (HMGB1) to form focal adhesions in cardiac fibroblasts; TGFβR activation via its SVVYGLR motif and subsequent induction of collagen-1 and -3 (Col1/3) expression; enhancement of Col1/3 expression via integrin receptor-mediated signaling pathways; inhibition of cardiac fibroblast apoptosis via blocking the release of cytochrome C (Cyt C) from mitochondria; inhibition of the activity and expression of MMP-2/9 induced by interleukin-1β (IL-1β) signaling pathway; induction of tissue inhibitor of metalloproteinases 1 (TIMP-1) expression via integrin- and CD44-mediated signaling pathways. The overall effect of osteopontin signaling in cardiac fibroblasts results in myofibroblast differentiation, proliferation, migration, and survival, synthesis of extracellular matrix (ECM) proteins, and modulation of ECM via regulating expression of MMPs and TIMPs.
Transforming growth factor-β (TGF-β) is a multifunctional cytokine mediating myofibroblast transformation and collagen synthesis. TGF-β1 failed to induce myofibroblast differentiation of osteopontin-depleted fibroblasts [
83], suggesting a pivotal role of osteopontin in myofibroblast differentiation. Osteopontin operates along with high mobility group box 1 intracellularly to form focal adhesions in the myofibroblasts in response to TGF-β1 stimulation [
83].
Osteopontin causes collagen-I synthesis and secretion in fibroblasts through a focal adhesion kinase (FAK)/protein kinase B (Akt)-dependent pathway [
86]. The thrombin-cleaved N-terminal osteopontin fragment can induce collagen-I and -III expression in cardiac fibroblasts via activation of the TGFβR signaling pathway [
85]. Ang-II-induced cardiac fibroblast proliferation and contraction are mediated by osteopontin RGD domain-β3-integrin receptor signaling pathway [
87]. A combination of osteopontin and Ang-II promoted the contraction of three-dimensional collagen gels and cardiac fibroblast growth via β3 integrin-mediated signaling pathways [
87].
Osteopontin inhibited the expression and activity of MMP-2 and MMP-9 in cardiac fibroblasts induced by IL-1β stimulation [
88]. These effects were mediated by β3 integrins-induced activation of the PKC-ζ signaling pathway [
88]. Osteopontin upregulated tissue inhibitor matrix metalloproteinase 1 (TIMP-1) and downregulated MMP-1 expression in fibroblasts via αvβ3 and CD44-receptor signaling [
89]. Remarkably, MMP-9-cleaved osteopontin fragments containing the RGD motif induced more pronounced cardiac fibroblast migration than the full-length osteopontin [
38].
Osteopontin is a strong regulator of lysyl oxidase expression and activity in cardiac fibroblasts, which is responsible for the formation of cross-linked, insoluble collagen and increased ECM accumulation and myocardial stiffness [
84,
90]. It induces lysyl oxidase upregulation in cardiac fibroblasts [
90] via increasing connective tissue growth factor expression [
83].
2.1.3. Osteopontin in Cardiac Endothelial Cells
Various growth factors and inflammatory mediators have been shown to regulate osteopontin expression in cardiac endothelial cells [
64,
65,
75] (
Figure 4). Ang-II increased osteopontin expression in cardiac endothelial cells [
65,
75] through NADPH-ROS-mediated activation of the Erk1/2 signaling pathway [
75]. A combination of IL-1β and IFN-γ also augmented osteopontin expression in cardiac endothelial cells [
64]. Moreover, dexamethasone significantly increased osteopontin expression in in vitro experiments [
53].
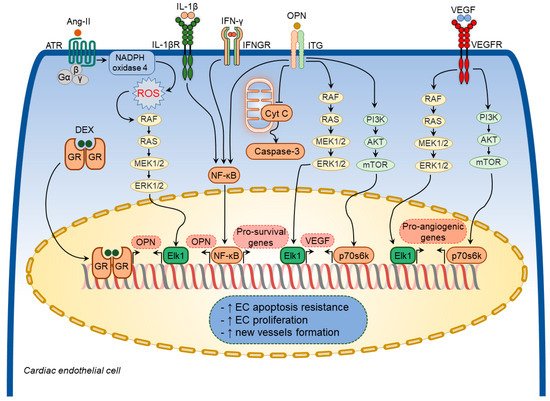
Figure 4. Osteopontin signaling in cardiac endothelial cells. A number of factors induce osteopontin (OPN) expression in cardiac endothelial cells including angiotensin-II (Ang-II), interleukin-1β (IL-1β), interferon-γ (IFN-γ), and dexamethasone (DEX). In turn, osteopontin regulates several cellular processes in cardiac endothelial cells via modulating certain signaling pathways. These pathways include osteopontin-induced vascular endothelial growth factor (VEGF) expression and subsequent VEGF-mediated expression of pro-angiogenic factors, inhibition of cardiac endothelial cell apoptosis via blocking mitochondria-mediated apoptosis pathway. Osteopontin signaling in cardiac endothelial cells results in increased apoptosis resistance, increased cell proliferation, and new vessels formation.
Osteopontin exerted cytoprotective effects on endothelial cells and promoted angiogenesis by enhancing vascular endothelial growth factor (VEGF) expression through the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)- and ERK-mediated pathways [
91]. In addition, osteopontin facilitated the survival of stressed endothelial cells [
92]. Endothelial cells plated on osteopontin-coated surfaces become apoptosis-resistant to serum deprivation via integrin-mediated activation of the nuclear factor kappa B (NF-κB) pathway [
93]. Similarly, soluble osteopontin inhibited apoptosis of endothelial cells deprived of essential growth factors [
92]. Osteopontin also promoted endothelial regeneration of the injured vessels through activation of RGD-αvβ3 signaling pathways [
94]. In line with these findings, osteopontin-null mice showed impaired myocardial angiogenic response post-MI, which resulted in adverse LV remodeling, suggesting that osteopontin plays a crucial role in maintaining and restoring myocardial capillarization following MI [
95]. Thus, osteopontin regulates coronary microvascular homeostasis by exerting cytoprotective and pro-survival effects on endothelial cells (
Figure 4).
2.2. Implications of Osteopontin in Specific Pathological Processes
2.2.1. Osteopontin in Cardiac Hypertrophy
Cardiac hypertrophy is an adaptive response to hemodynamic load characterized by the thickening of the heart walls and enlargement of the individual cardiomyocytes, which has a compensatory role in maintaining cardiac performance and attenuating ventricular wall stress and chamber dilatation. At the cellular level, cardiomyocyte hypertrophy is characterized by an increase in cell dimensions mainly due to augmented contractile protein synthesis [
101]. However, sustained hemodynamic stress in pathological conditions such as hypertension and valvular heart disease drives adverse alterations in the myocardium characterized by excessive cardiomyocyte apoptosis, myocardial inflammation, and fibrosis, eventually culminating in ventricular dilation and dysfunction.
Robust upregulation of myocardial osteopontin expression upon mechanical stress was clearly shown in several rodent models [
58,
68,
85]. Pressure overload induced an acute and strong elevation of osteopontin expression in the myocardium in rodents following TAC [
58,
68,
85] and in rats with renovascular hypertension [
58]. The increase in myocardial osteopontin expression was reported to be proportional to the degree of the afterload [
102]. In line with these findings, there was a strong correlation between expression levels of osteopontin and atrial natriuretic peptide in the LV myocardium in renovascular and TAC-induced LV remodeling models [
58]. In the latter models, cardiomyocytes were the main cell type in the heart producing osteopontin [
58]. In contrast, in spontaneously hypertensive rats with cardiac hypertrophy, osteopontin expression was identified primarily in non-myocytes in the interstitial and perivascular space (possibly infiltrating macrophages and fibroblasts) [
103].
The exploitation of osteopontin-null mice in several animal models of cardiac hypertrophy revealed that osteopontin is a key mediator in the mechanical stress-induced myocardial hypertrophic response [
59,
68,
76,
80]. In mice subjected to TAC, a lack of osteopontin was associated with attenuated cardiac hypertrophic response [
68]. Knocking out osteopontin in transgenic mice with NHE1 overexpression significantly reduced cardiac hypertrophy, attenuated collagen deposition, and improved cardiac function [
76]. Similarly, in another genetic mouse model of HF with preserved ejection fraction induced by Col4a3 deficiency, osteopontin deletion was associated with improved parameters of LV diastolic function and cardiac hypertrophy reduction [
80]. Controversial data were obtained in the model of Ang-II infusion [
29,
59]. Thus, in one study, osteopontin deficiency attenuated cardiac hypertrophy induced by Ang-II infusion [
59], whereas, in another study, osteopontin deletion did not prevent cardiac hypertrophy, despite attenuated myocardial fibrosis [
29].
2.2.2. Osteopontin in Cardiac Inflammation
Heart diseases are associated with inflammation, which contributes to cardiac dysfunction and myocardial fibrosis [
10,
104,
105]. Numerous cytokines and chemokines are actively involved in the onset and progression of myocardial fibrosis [
10,
105]. Osteopontin has been shown to exert cytokine- and chemokine-like functions [
35].
Among various inflammatory cells, macrophages represent the dominant inflammatory cell population in the remodeled myocardium [
106]. Macrophage infiltration of the myocardium has been shown in various models, including spontaneously hypertensive rats and murine TAC [
106,
107]. Complete abolition of CD45+ monocytes recruitment to the myocardium in response to Ang-II infusion in osteopontin-null mice [
108] suggested the involvement of osteopontin in this process.
Osteopontin expressed in circulating leucocytes may also be actively involved in inflammatory cell recruitment to the myocardium and systemic inflammation in patients with various cardiac diseases [
109]. Circulating CD4+ T lymphocytes expressing osteopontin and circulating osteopontin levels both correlate with the New York Heart Association Functional Class (NYHA FC) in HF patients and are associated with plaque instability in coronary artery disease patients [
109,
110].
Inflammatory cells have been identified as one of the major contributors to fibrogenesis in different tissues and organs under various disease conditions [
111]. During the acute inflammatory phase of wound healing processes upon tissue injury in different tissues and organs, osteopontin expression is enhanced in infiltrating leukocytes, while during the chronic inflammatory phase, its upregulation is observed in resident macrophages [
34]. Increased osteopontin expression by resident and recruited inflammatory cells increases collagen deposition and accumulation in the tissues [
109,
110,
112].
Osteopontin expression by immune/inflammatory cells is associated with cardiac hypertrophic and fibrotic responses in the settings of a number of heart diseases [
62,
63,
69,
108,
113]. Studies exploiting mice with a genetically modified osteopontin expression provided important insights into its role in myocardial inflammation in diverse cardiac conditions [
69,
79,
112,
114,
115,
116,
117]. Mice overexpressing osteopontin in cardiomyocytes spontaneously develop severe cardiomyopathy, which is characterized by enhanced recruitment of inflammatory cells to the myocardium and excessive collagen accumulation, subsequently leading to chronic myocarditis and eventually premature death [
79,
112]. In line with these reports, deletion of osteopontin in desmin deficient mice, which spontaneously develop DCM with increased myocardial inflammation and fibrosis, was associated with an attenuation of myocardial inflammation and improvement in LV systolic function [
69].
Cardiomyocyte-specific integrin-linked kinase (ILK) deficient mice spontaneously develop lethal cardiomyopathy characterized by excessive inflammatory cell accumulation, myocardial fibrosis, and cardiomyocyte apoptosis [
115]. Augmented osteopontin expression in cardiomyocytes revealed by comprehensive profiling and mitigation of HF severity in these mice by application of anti-osteopontin antibodies implicated osteopontin as a major contributor to this phenotype [
115]. Likewise, in other murine models, including those of Chagas heart disease and viral myocarditis, osteopontin-null mice displayed improved cardiac remodeling and attenuated myocardial inflammation [
116] [
117].
When taken together, osteopontin is highly upregulated in acute and chronic inflammatory phases of the myocardial injury and can contribute to adverse myocardial remodeling and cardiac dysfunction by promoting the recruitment of inflammatory cells to the myocardium.
2.3. Clinical Implication of Osteopontin
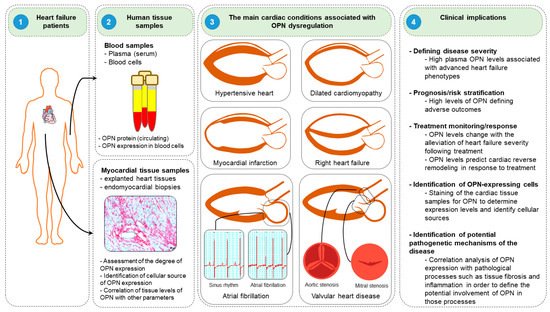
Clinical studies have suggested that osteopontin might serve as a potent diagnostic and prognostic biomarker in diverse HF conditions. In this section, we discuss the roles and clinical implications of osteopontin in various HF diseases, including DCM, hypertensive HF, MI, and right HF (Figure 5).
Figure 5. Clinical implications of osteopontin. (1) Blood samples are collected from the peripheral blood, while myocardial tissues are obtained from explanted hearts or endomyocardial biopsies during cardiac catheterization or cardiac surgery. (2) Plasma/serum and blood cells are isolated from blood samples to measure osteopontin (OPN) levels. Similarly, osteopontin expression levels are evaluated and cellular sources of osteopontin are identified in myocardial tissues. (3) Assessment of osteopontin levels might be useful in several cardiac conditions, including hypertensive heart, dilated cardiomyopathy, myocardial infarction, and right heart failure, atrial fibrillation and valvular heart disease. (4) Assessment of osteopontin levels might be useful in determining diseases severity, prognostic/risk stratification, monitoring of treatment response, identification of cellular sources of osteopontin in various cardiac conditions and help to understand the mechanisms of the disease pathogenesis.