Glioma are the most frequent malignant primary CNS tumors in adults, with an incidence of 5–6 per 100,000 per year, with glioblastoma (with 3.2 per 100,000 per year) being the largest subgroup. The current therapy for glioblastoma is resection followed by radiochemotherapy and their prognosis is always fatal.Oncogenic fusion genes emerged as successful targets in several malignancies, such as chronic myeloic leukemia or lung cancer. Fusion of the fibroblast growth receptor 3 and the transforming acidic coiled coil containing protein – FGFR3-TACC3-fusion is prevalent in 3-4% of human glioblastoma. The fusion protein leads to constitutively activated kinase signaling of FGFR3 and thereby promotes cell proliferation and tumour progression. An overview on clinical and histomolecular features of FGFR3-TACC3-fusion positive glioblastoma is described and the cellular fuction of the fusion protein in glioblastoma cells is highlighted.
- FGFR3-TACC3
- glioblastoma
- tumor cells
1. The FGFR Family and FGFR Signaling
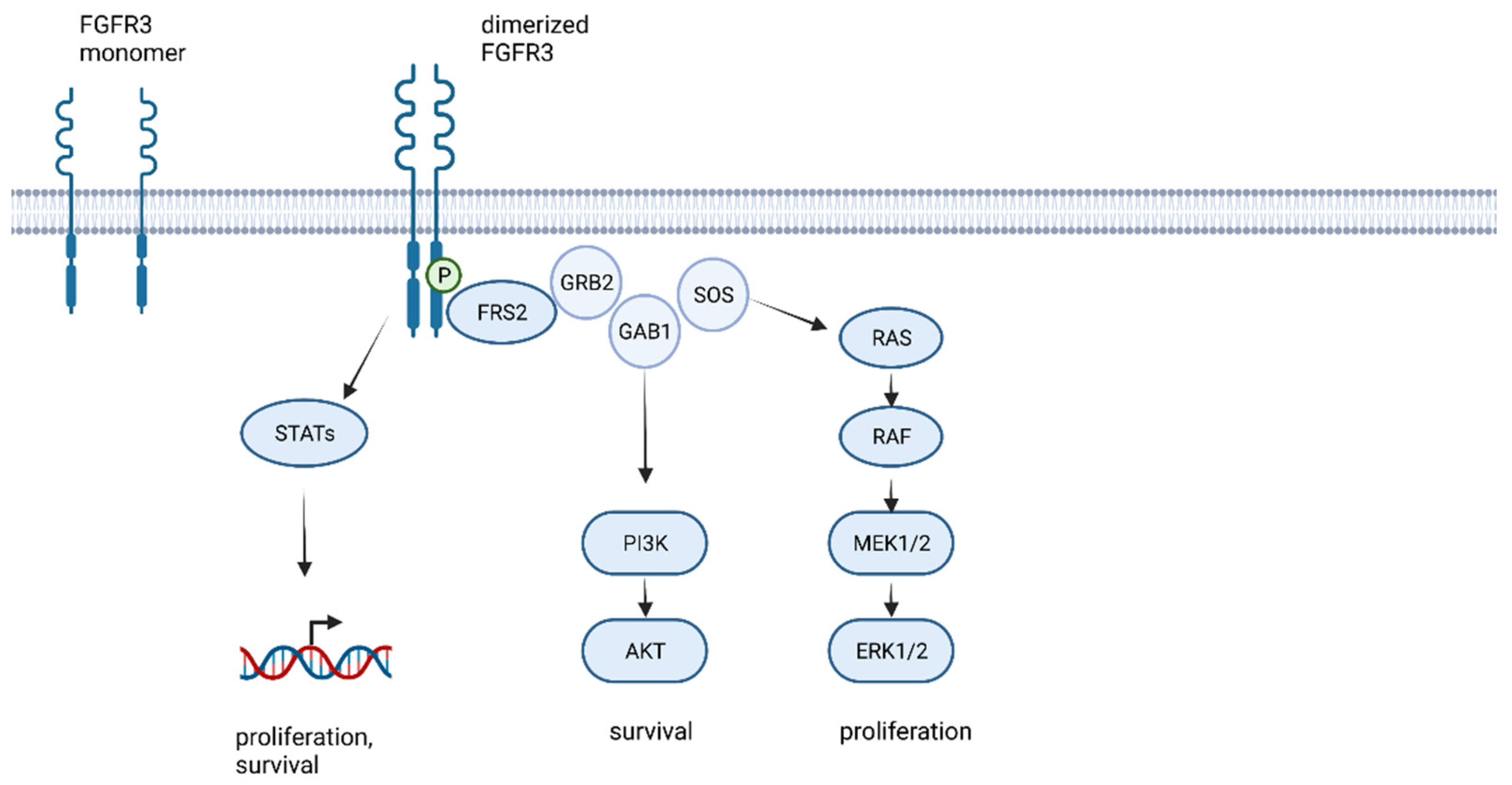
2. Histomolecular and Clinical Characteristics of FGFR3-TACC3 Fused Glioblastoma
3. The Functional Role of FGFR3-TACC3 in Glioblastoma Cells
This entry is adapted from the peer-reviewed paper 10.3390/ijms23158675
References
- Harmer, N.J.; Ilag, L.L.; Mulloy, B.; Pellegrini, L.; Robinson, C.V.; Blundell, T.L. Towards a resolution of the stoichiometry of the fibroblast growth factor (FGF)-FGF receptor-heparin complex. J. Mol. Biol. 2004, 339, 821–834.
- Williams, S.V.; Hurst, C.D.; Knowles, M.A. Oncogenic FGFR3 gene fusions in bladder cancer. Hum. Mol. Genet. 2013, 22, 795–803.
- Warzecha, C.C.; Sato, T.K.; Nabet, B.; Hogenesch, J.B.; Carstens, R.P. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell 2009, 33, 591–601.
- Latko, M.; Czyrek, A.; Porębska, N.; Kucińska, M.; Otlewski, J.; Zakrzewska, M.; Opaliński, Ł. Cross-Talk between Fibroblast Growth Factor Receptors and Other Cell Surface Proteins. Cells 2019, 8, 455.
- Kucińska, M.; Porębska, N.; Lampart, A.; Latko, M.; Knapik, A.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Differential regulation of fibroblast growth factor receptor 1 trafficking and function by extracellular galectins. Cell Commun. Signal. 2019, 17, 65.
- Parker, B.C.; Engels, M.; Annala, M.; Zhang, W. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J. Pathol. 2014, 232, 4–15.
- Jimenez-Pascual, A.; Siebzehnrubl, F.A. Fibroblast Growth Factor Receptor Functions in Glioblastoma. Cells 2019, 8, 715.
- Costa, R.; Carneiro, B.A.; Taxter, T.; Tavora, F.A.; Kalyan, A.; Pai, S.A.; Chae, Y.K.; Giles, F.J. FGFR3-TACC3 fusion in solid tumors: Mini review. Oncotarget 2016, 7, 55924–55938.
- Bae, J.H.; Boggon, T.J.; Tomé, F.; Mandiyan, V.; Lax, I.; Schlessinger, J. Asymmetric receptor contact is required for tyrosine autophosphorylation of fibroblast growth factor receptor in living cells. Proc. Natl. Acad. Sci. USA 2010, 107, 2866–2871.
- Ferguson, H.R.; Smith, M.P.; Francavilla, C. Fibroblast Growth Factor Receptors (FGFRs) and Noncanonical Partners in Cancer Signaling. Cells 2021, 10, 1201.
- Liang, G.; Liu, Z.; Wu, J.; Cai, Y.; Li, X. Anticancer molecules targeting fibroblast growth factor receptors. Trends Pharmacol. Sci. 2012, 33, 531–541.
- Parker, B.C.; Annala, M.J.; Cogdell, D.E.; Granberg, K.J.; Sun, Y.; Ji, P.; Li, X.; Gumin, J.; Zheng, H.; Hu, L.; et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J. Clin. Invest. 2013, 123, 855–865.
- Lo, T.L.; Yusoff, P.; Fong, C.W.; Guo, K.; McCaw, B.J.; Phillips, W.A.; Yang, H.; Wong, E.S.M.; Leong, H.F.; Zeng, Q.; et al. The ras/mitogen-activated protein kinase pathway inhibitor and likely tumor suppressor proteins, sprouty 1 and sprouty 2 are deregulated in breast cancer. Cancer Res. 2004, 64, 6127–6136.
- Yusoff, P.; Lao, D.-H.; Ong, S.H.; Wong, E.S.M.; Lim, J.; Lo, T.L.; Leong, H.F.; Fong, C.W.; Guy, G.R. Sprouty2 inhibits the Ras/MAP kinase pathway by inhibiting the activation of Raf. J. Biol. Chem. 2002, 277, 3195–3201.
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic. Acids Res. 2017, 45, D777–D783.
- Gouazé-Andersson, V.; Delmas, C.; Taurand, M.; Martinez-Gala, J.; Evrard, S.; Mazoyer, S.; Toulas, C.; Cohen-Jonathan-Moyal, E. FGFR1 Induces Glioblastoma Radioresistance through the PLCγ/Hif1α Pathway. Cancer Res. 2016, 76, 3036–3044.
- Jimenez-Pascual, A.; Hale, J.S.; Kordowski, A.; Pugh, J.; Silver, D.J.; Bayik, D.; Roversi, G.; Alban, T.J.; Rao, S.; Chen, R.; et al. ADAMDEC1 Maintains a Growth Factor Signaling Loop in Cancer Stem Cells. Cancer Discov. 2019, 9, 1574–1589.
- Wang, Z.; Zhang, C.; Sun, L.; Liang, J.; Liu, X.; Li, G.; Yao, K.; Zhang, W.; Jiang, T. FGFR3, as a receptor tyrosine kinase, is associated with differentiated biological functions and improved survival of glioma patients. Oncotarget 2016, 7, 84587–84593.
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235.
- Ferguson, S.D.; Zhou, S.; Huse, J.T.; de Groot, J.F.; Xiu, J.; Subramaniam, D.S.; Mehta, S.; Gatalica, Z.; Swensen, J.; Sanai, N.; et al. Targetable Gene Fusions Associate with the IDH Wild-Type Astrocytic Lineage in Adult Gliomas. J. Neuropathol. Exp. Neurol. 2018, 77, 437–442.
- Mata, D.A.; Benhamida, J.K.; Lin, A.L.; Vanderbilt, C.M.; Yang, S.-R.; Villafania, L.B.; Ferguson, D.C.; Jonsson, P.; Miller, A.M.; Tabar, V.; et al. Genetic and epigenetic landscape of IDH-wildtype glioblastomas with FGFR3-TACC3 fusions. Acta Neuropathol. Commun. 2020, 8, 186.
- Di Stefano, A.L.; Picca, A.; Saragoussi, E.; Bielle, F.; Ducray, F.; Villa, C.; Eoli, M.; Paterra, R.; Bellu, L.; Mathon, B.; et al. Clinical, molecular, and radiomic profile of gliomas with FGFR3-TACC3 fusions. Neuro-oncology 2020, 22, 1614–1624.
- Bielle, F.; Di Stefano, A.-L.; Meyronet, D.; Picca, A.; Villa, C.; Bernier, M.; Schmitt, Y.; Giry, M.; Rousseau, A.; Figarella-Branger, D.; et al. Diffuse gliomas with FGFR3-TACC3 fusion have characteristic histopathological and molecular features. Brain Pathol. 2018, 28, 674–683.
- Di Stefano, A.L.; Fucci, A.; Frattini, V.; Labussiere, M.; Mokhtari, K.; Zoppoli, P.; Marie, Y.; Bruno, A.; Boisselier, B.; Giry, M.; et al. Detection, Characterization, and Inhibition of FGFR-TACC Fusions in IDH Wild-type Glioma. Clin. Cancer Res. 2015, 21, 3307–3317.
- Granberg, K.J.; Annala, M.; Lehtinen, B.; Kesseli, J.; Haapasalo, J.; Ruusuvuori, P.; Yli-Harja, O.; Visakorpi, T.; Haapasalo, H.; Nykter, M.; et al. Strong FGFR3 staining is a marker for FGFR3 fusions in diffuse gliomas. Neuro-oncology 2017, 19, 1206–1216.
- Schittenhelm, J.; Ziegler, L.; Sperveslage, J.; Mittelbronn, M.; Capper, D.; Burghardt, I.; Poso, A.; Biskup, S.; Skardelly, M.; Tabatabai, G. FGFR3 overexpression is a useful detection tool for FGFR3 fusions and sequence variations in glioma. Neurooncol. Pract. 2021, 8, 209–221.
- Broggi, G.; Piombino, E.; Altieri, R.; Romano, C.; Certo, F.; Barbagallo, G.M.V.; Vigneri, P.; Condorelli, D.; Colarossi, L.; Colarossi, C.; et al. Glioblastoma, IDH-Wild Type with FGFR3-TACC3 Fusion: When Morphology May Reliably Predict the Molecular Profile of a Tumor. A Case Report and Literature Review. Front. Neurol. 2022, 13, 823015.
- Gilani, A.; Davies, K.D.; Kleinschmidt-DeMasters, B.K. Can adult IDH-wildtype glioblastomas with FGFR3:TACC3 fusions be reliably predicted by histological features? Clin. Neuropathol. 2021, 40, 165–167.
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-oncology 2020, 22, iv1–iv96.
- Wu, Z.; Lopes Abath Neto, O.; Bale, T.A.; Benhamida, J.; Mata, D.; Turakulov, R.; Abdullaev, Z.; Marker, D.; Ketchum, C.; Chung, H.-J.; et al. DNA methylation analysis of glioblastomas harboring FGFR3-TACC3 fusions identifies a methylation subclass with better patient survival. Acta Neuropathol. 2022, 144, 155–157.
- Vagvala, S.; Guenette, J.P.; Jaimes, C.; Huang, R.Y. Imaging diagnosis and treatment selection for brain tumors in the era of molecular therapeutics. Cancer Imaging 2022, 22, 19.
- Wu, Y.-M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.-F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647.
- Nelson, K.N.; Meyer, A.N.; Wang, C.G.; Donoghue, D.J. Oncogenic driver FGFR3-TACC3 is dependent on membrane trafficking and ERK signaling. Oncotarget 2018, 9, 34306–34319.
- Nelson, K.N.; Meyer, A.N.; Siari, A.; Campos, A.R.; Motamedchaboki, K.; Donoghue, D.J. Oncogenic Gene Fusion FGFR3-TACC3 Is Regulated by Tyrosine Phosphorylation. Mol. Cancer Res. 2016, 14, 458–469.
- Frattini, V.; Pagnotta, S.M.; Tala; Fan, J.J.; Russo, M.V.; Lee, S.B.; Garofano, L.; Zhang, J.; Shi, P.; Lewis, G.; et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature 2018, 553, 222–227.
- Daly, C.; Castanaro, C.; Zhang, W.; Zhang, Q.; Wei, Y.; Ni, M.; Young, T.M.; Zhang, L.; Burova, E.; Thurston, G. FGFR3-TACC3 fusion proteins act as naturally occurring drivers of tumor resistance by functionally substituting for EGFR/ERK signaling. Oncogene 2017, 36, 471–481.
- Lombardi, B.; Ashford, P.; Moya-Garcia, A.A.; Rust, A.; Crawford, M.; Williams, S.V.; Knowles, M.A.; Katan, M.; Orengo, C.; Godovac-Zimmermann, J. Unique signalling connectivity of FGFR3-TACC3 oncoprotein revealed by quantitative phosphoproteomics and differential network analysis. Oncotarget 2017, 8, 102898–102911.