Despite increasing availability and more successful interventional approaches to restore coronary reperfusion, myocardial ischemia-reperfusion injury is a substantial cause of morbidity and mortality worldwide. During myocardial ischemia, the myocardium becomes profoundly hypoxic, thus causing stabilization of hypoxia-inducible transcription factors (HIF). Stabilization of HIF leads to a transcriptional program that promotes adaptation to hypoxia and cellular survival. Transcriptional consequences of HIF stabilization include increases in extracellular production and signaling effects of adenosine. Extracellular adenosine functions as a signaling molecule via the activation of adenosine receptors. Several studies implicated adenosine signaling in cardioprotection, particularly through the activation of the Adora2a and Adora2b receptors. Adenosine receptor activation can lead to metabolic adaptation to enhance ischemia tolerance or dampen myocardial reperfusion injury via signaling events on immune cells. Many studies highlight that clinical strategies to target the hypoxia-adenosine link could be considered for clinical trials. This could be achieved by using pharmacologic HIF activators or by directly enhancing extracellular adenosine production or signaling as a therapy for patients with acute myocardial infarction, or undergoing cardiac surgery.
- adenosine
- hypoxia
- CD73
- CD39
- Adora2a
- A2A
- A2B
- Adora2b
- ENT1
- ENT2
1. Hypoxia-Inducible Transcription Factors (HIF) Are Stabilized during Myocardial Ischemia and Provide Cardioprotection
2. Role of HIF in Regulating Adenosine Signaling during Myocardial Ischemia-Reperfusion Injury
2.1. Impact of Hypoxia-Signaling on the Production of Extracellular Adenosine
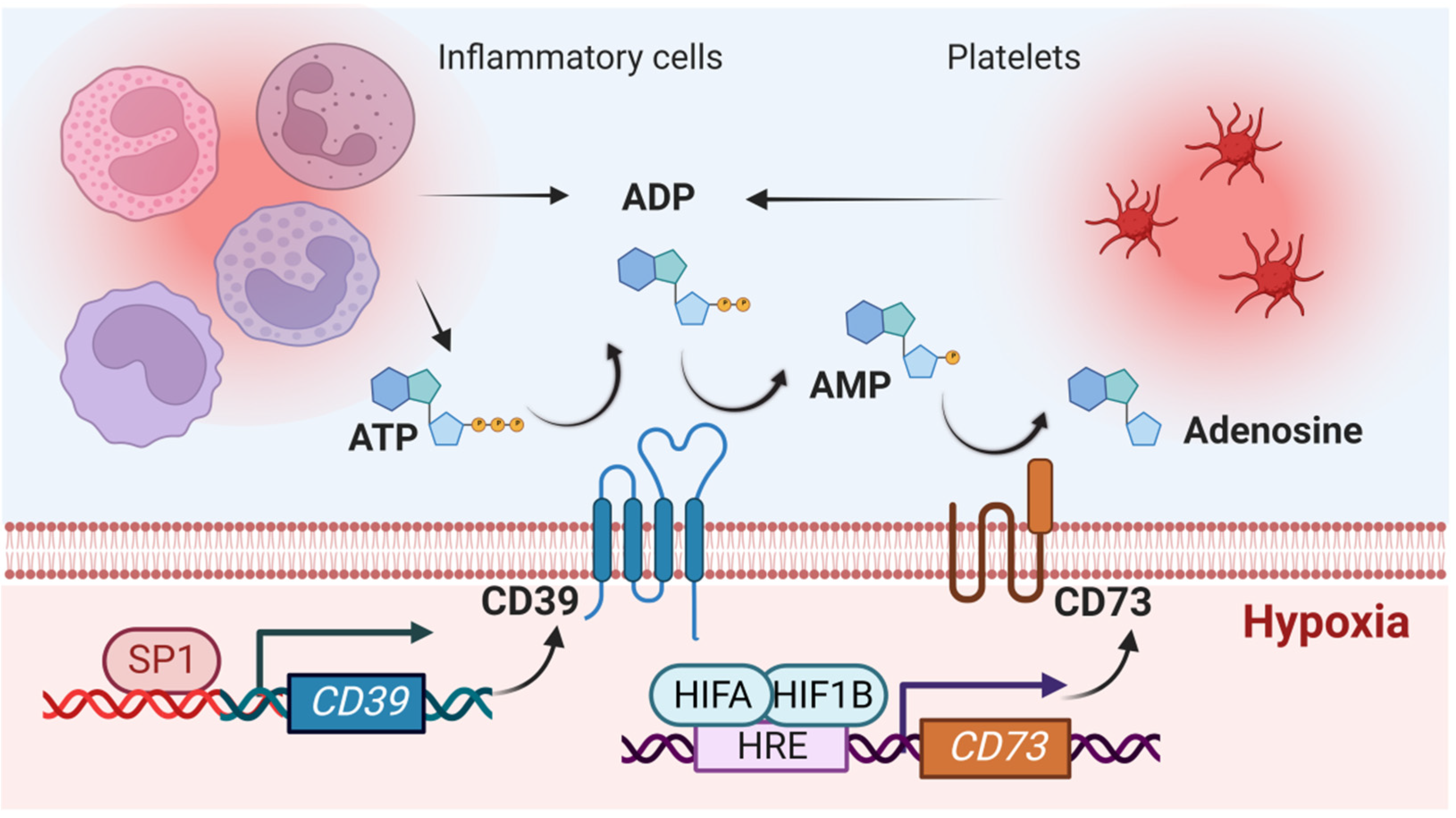
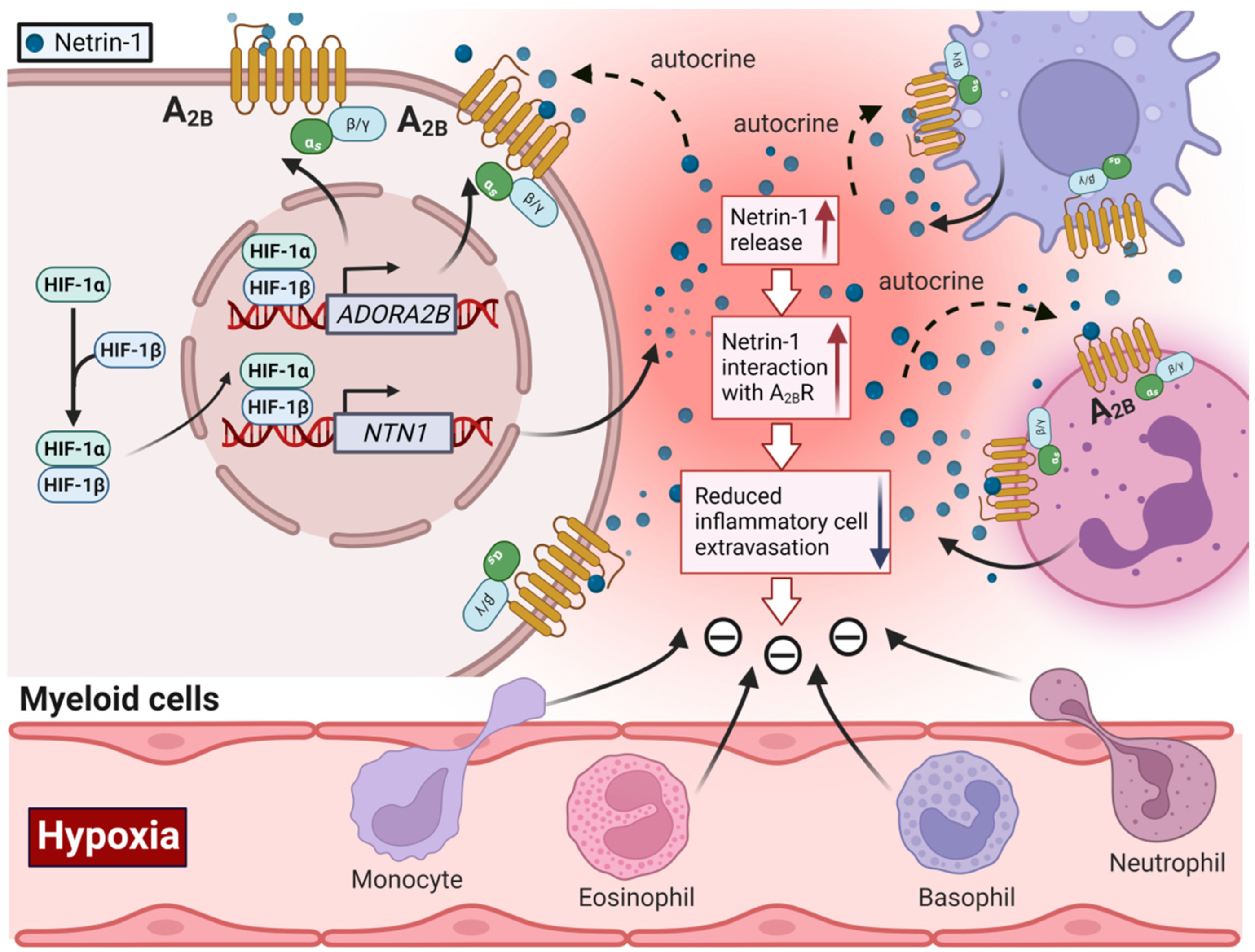
2.2. Role of HIF in Coordinating Extracellular Adenosine Signaling during Myocardial Ischemia-Reperfusion Injury
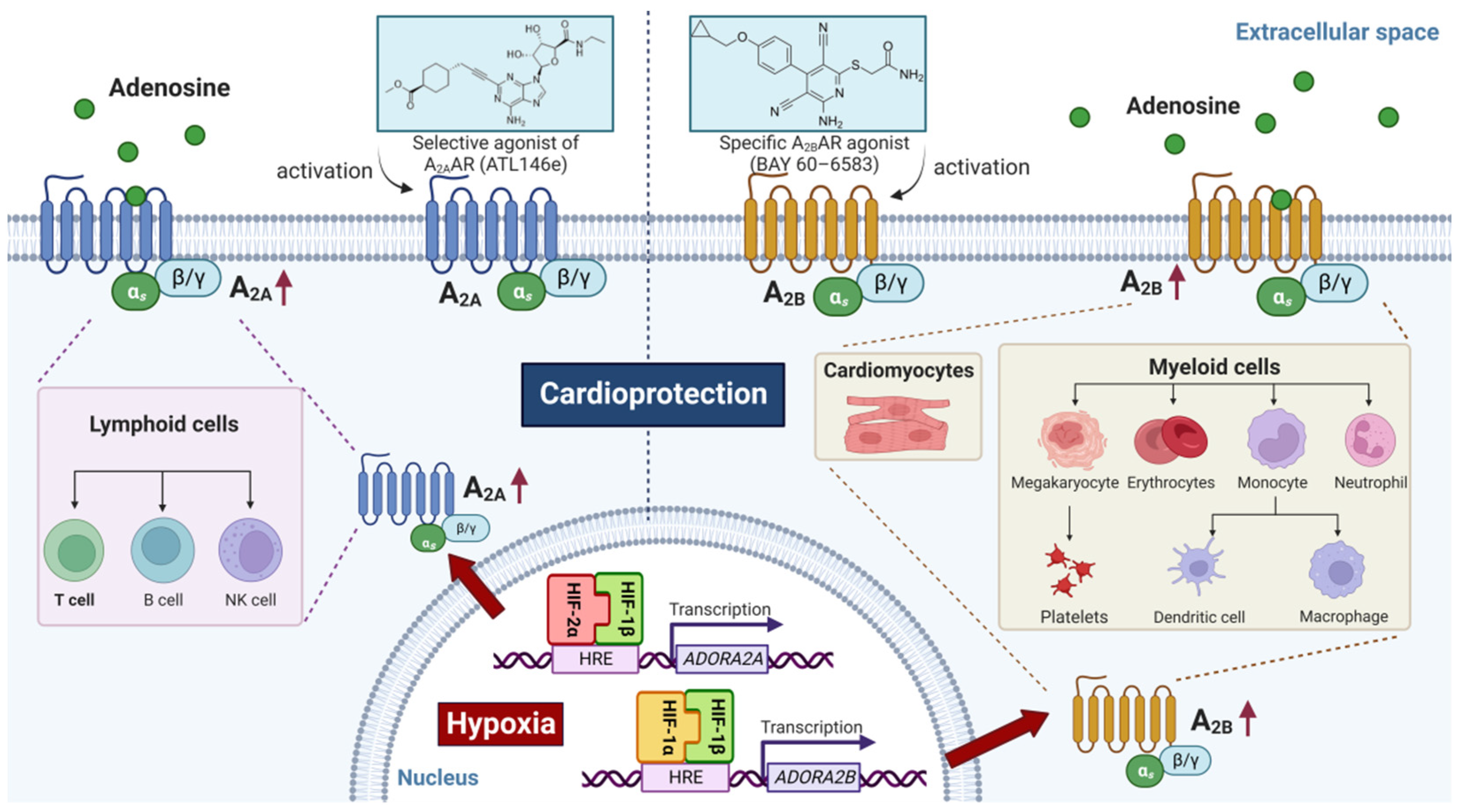
2.3. HIF-Dependent Promotion of Alternative Adenosine Receptor Activation
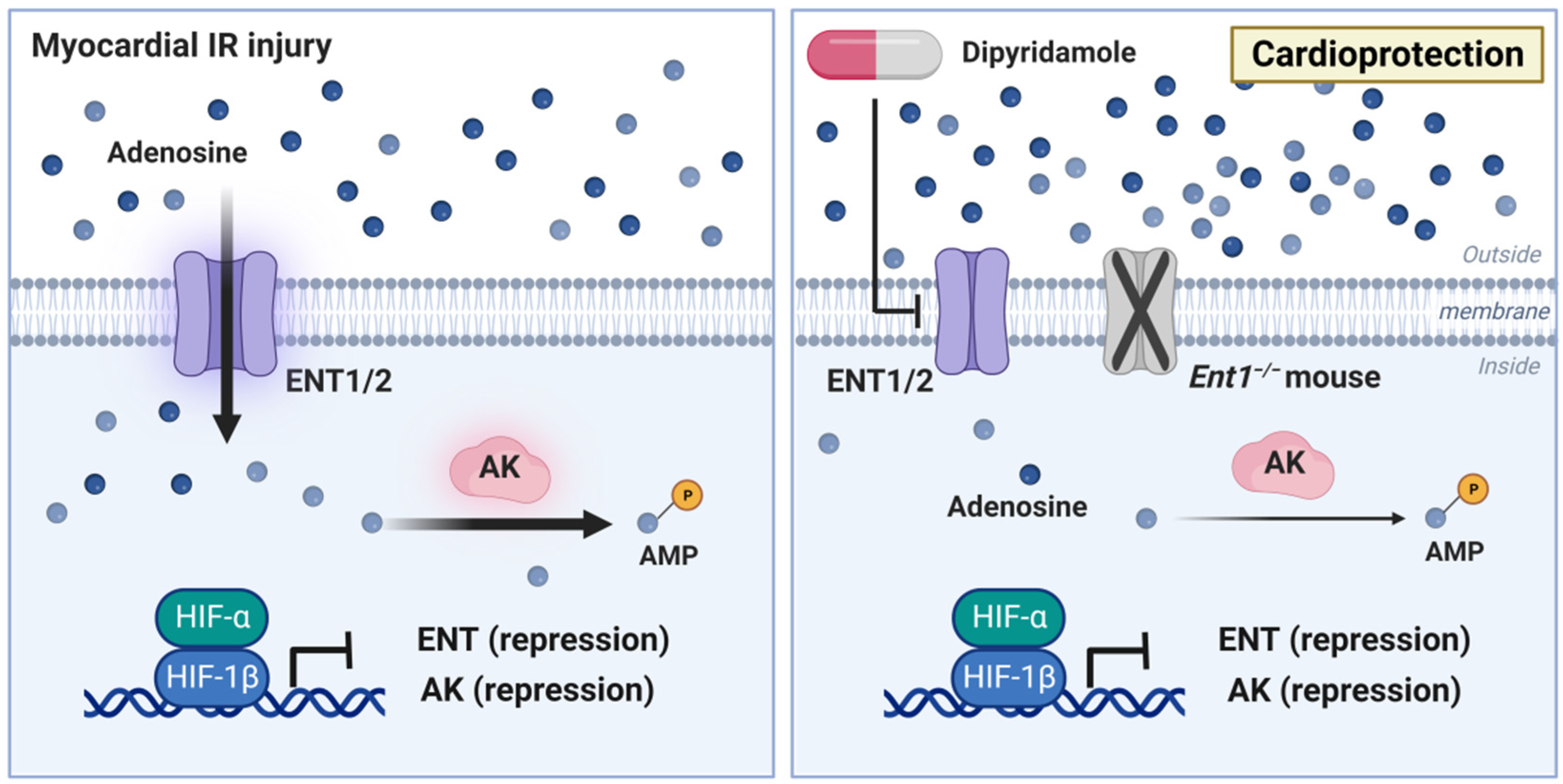
2.4. Impact of HIF Signaling on Extracellular Adenosine Uptake and Metabolism
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10081939
References
- Eckle, T.; Kohler, D.; Lehmann, R.; El Kasmi, K.C.; Eltzschig, H.K. Hypoxia-Inducible Factor-1 Is Central to Cardioprotection: A New Paradigm for Ischemic Preconditioning. Circulation 2008, 118, 166–175.
- Wang, G.; Jiang, B.; Rue, E.; Semenza, G. Hypoxia-Inducible Factor 1 is a Basic-Helix-Loop-Helix-PAS Heterodimer Regulated by Cellular O2 Tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514.
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763.
- Wang, G.L.; Semenza, G.L. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J. Biol. Chem. 1993, 268, 21513–21518.
- Colgan, S.P.; Furuta, G.T.; Taylor, C.T. Hypoxia and Innate Immunity: Keeping Up with the HIFsters. Annu. Rev. Immunol. 2020, 38, 341–363.
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665.
- Li, X.; Berg, N.K.; Mills, T.; Zhang, K.; Eltzschig, H.K.; Yuan, X. Adenosine at the Interphase of Hypoxia and Inflammation in Lung Injury. Front. Immunol. 2020, 11, 604944.
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019, 51, 1–13.
- Yuan, X.; Lee, J.W.; Bowser, J.L.; Neudecker, V.; Sridhar, S.; Eltzschig, H.K. Targeting Hypoxia Signaling for Perioperative Organ Injury. Anesth. Analg. 2018, 126, 308–321.
- Taylor, C.T.; Colgan, S.P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 2017, 17, 774–785.
- Fraisl, P.; Aragones, J.; Carmeliet, P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat. Rev. Drug Discov. 2009, 8, 139–152.
- Brown, E.; Rowan, C.; Strowitzki, M.J.; Fagundes, R.R.; Faber, K.N.; Guntsch, A.; Halligan, D.N.; Kugler, J.; Jones, F.; Lee, C.T.; et al. Mucosal inflammation downregulates PHD1 expression promoting a barrier-protective HIF-1alpha response in ulcerative colitis patients. FASEB J. 2020, 34, 3732–3742.
- Kennel, K.B.; Burmeister, J.; Schneider, M.; Taylor, C.T. The PHD1 oxygen sensor in health and disease. J. Physiol. 2018, 596, 3899–3913.
- Tambuwala, M.M.; Cummins, E.P.; Lenihan, C.R.; Kiss, J.; Stauch, M.; Scholz, C.C.; Fraisl, P.; Lasitschka, F.; Mollenhauer, M.; Saunders, S.P.; et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology 2010, 139, 2093–2101.
- Bowser, J.L.; Phan, L.H.; Eltzschig, H.K. The Hypoxia-Adenosine Link during Intestinal Inflammation. J. Immunol. 2018, 200, 897–907.
- Koeppen, M.; Eckle, T.; Eltzschig, H.K. Interplay of hypoxia and A2B adenosine receptors in tissue protection. Adv. Pharmacol. 2011, 61, 145–186.
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732.
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671.
- Cartwright, I.M.; Dowdell, A.S.; Lanis, J.M.; Brink, K.R.; Mu, A.; Kostelecky, R.E.; Schaefer, R.E.M.; Welch, N.; Onyiah, J.C.; Hall, C.H.T.; et al. Mucosal acidosis elicits a unique molecular signature in epithelia and intestinal tissue mediated by GPR31-induced CREB phosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2023871118.
- Vohwinkel, C.U.; Coit, E.J.; Burns, N.; Elajaili, H.; Hernandez-Saavedra, D.; Yuan, X.; Eckle, T.; Nozik, E.; Tuder, R.M.; Eltzschig, H.K. Targeting alveolar-specific succinate dehydrogenase A attenuates pulmonary inflammation during acute lung injury. FASEB J. 2021, 35, e21468.
- Eckle, T.; Brodsky, K.; Bonney, M.; Packard, T.; Han, J.; Borchers, C.H.; Mariani, T.J.; Kominsky, D.J.; Mittelbronn, M.; Eltzschig, H.K. HIF1A reduces acute lung injury by optimizing carbohydrate metabolism in the alveolar epithelium. PLoS Biol. 2013, 11, e1001665.
- Eltzschig, H.K.; Bratton, D.L.; Colgan, S.P. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat. Rev. Drug Discov. 2014, 13, 852–869.
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669.
- Bowser, J.L.; Lee, J.W.; Yuan, X.; Eltzschig, H.K. The Hypoxia-Adenosine Link during Inflammation. J. Appl. Physiol. 2017, 123, 1303–1320.
- Berg, N.K.; Li, J.; Kim, B.; Mills, T.; Pei, G.; Zhao, Z.; Li, X.; Zhang, X.; Ruan, W.; Eltzschig, H.K.; et al. Hypoxia-inducible factor-dependent induction of myeloid-derived netrin-1 attenuates natural killer cell infiltration during endotoxin-induced lung injury. FASEB J. 2021, 35, e21334.
- Eltzschig, H.K.; Abdulla, P.; Hoffman, E.; Hamilton, K.E.; Daniels, D.; Schonfeld, C.; Loffler, M.; Reyes, G.; Duszenko, M.; Karhausen, J.; et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J. Exp. Med. 2005, 202, 1493–1505.
- Zheng, W.; Kuhlicke, J.; Jackel, K.; Eltzschig, H.K.; Singh, A.; Sjoblom, M.; Riederer, B.; Weinhold, C.; Seidler, U.; Colgan, S.P.; et al. Hypoxia inducible factor-1 (HIF-1)-mediated repression of cystic fibrosis transmembrane conductance regulator (CFTR) in the intestinal epithelium. FASEB J. 2009, 23, 204–213.
- Morote-Garcia, J.C.; Rosenberger, P.; Nivillac, N.M.; Coe, I.R.; Eltzschig, H.K. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology 2009, 136, 607–618.
- Bruning, U.; Cerone, L.; Neufeld, Z.; Fitzpatrick, S.F.; Cheong, A.; Scholz, C.C.; Simpson, D.A.; Leonard, M.O.; Tambuwala, M.M.; Cummins, E.P.; et al. MicroRNA-155 promotes resolution of hypoxia-inducible factor 1alpha activity during prolonged hypoxia. Mol. Cell. Biol. 2011, 31, 4087–4096.
- Ju, C.; Wang, M.; Tak, E.; Kim, B.; Emontzpohl, C.; Yang, Y.; Yuan, X.; Kutay, H.; Liang, Y.; Hall, D.R.; et al. Hypoxia-inducible factor-1alpha-dependent induction of miR122 enhances hepatic ischemia tolerance. J. Clin. Investig. 2021, 131, e140300.
- Koeppen, M.; Lee, J.W.; Seo, S.W.; Brodsky, K.S.; Kreth, S.; Yang, I.V.; Buttrick, P.M.; Eckle, T.; Eltzschig, H.K. Hypoxia-inducible factor 2-alpha-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury. Nat. Commun. 2018, 9, 816.
- Eltzschig, H.K.; Bonney, S.K.; Eckle, T. Attenuating myocardial ischemia by targeting A2B adenosine receptors. Trends Mol. Med. 2013, 19, 345–354.
- Lee, J.W.; Koeppen, M.; Seo, S.W.; Bowser, J.L.; Yuan, X.; Li, J.; Sibilia, M.; Ambardekar, A.V.; Zhang, X.; Eckle, T.; et al. Transcription-independent Induction of ERBB1 through Hypoxia-inducible Factor 2A Provides Cardioprotection during Ischemia and Reperfusion. Anesthesiology 2020, 132, 763–780.
- Gao, R.Y.; Wang, M.; Liu, Q.; Feng, D.; Wen, Y.; Xia, Y.; Colgan, S.P.; Eltzschig, H.K.; Ju, C. Hypoxia-Inducible Factor-2alpha Reprograms Liver Macrophages to Protect Against Acute Liver Injury Through the Production of Interleukin-6. Hepatology 2020, 71, 2105–2117.
- Eckle, T.; Hartmann, K.; Bonney, S.; Reithel, S.; Mittelbronn, M.; Walker, L.A.; Lowes, B.D.; Han, J.; Borchers, C.H.; Buttrick, P.M.; et al. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat. Med. 2012, 18, 774–782.
- Chertow, G.M.; Pergola, P.E.; Farag, Y.M.K.; Agarwal, R.; Arnold, S.; Bako, G.; Block, G.A.; Burke, S.; Castillo, F.P.; Jardine, A.G.; et al. Vadadustat in Patients with Anemia and Non-Dialysis-Dependent CKD. N. Engl. J. Med. 2021, 384, 1589–1600.
- Kaplan, J. Roxadustat and Anemia of Chronic Kidney Disease. N. Engl. J. Med. 2019, 381, 1070–1072.
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2019, 381, 1001–1010.
- Chen, N.; Hao, C.; Liu, B.C.; Lin, H.; Wang, C.; Xing, C.; Liang, X.; Jiang, G.; Liu, Z.; Li, X.; et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N. Engl. J. Med. 2019, 381, 1011–1022.
- Kiers, D.; Wielockx, B.; Peters, E.; van Eijk, L.T.; Gerretsen, J.; John, A.; Janssen, E.; Groeneveld, R.; Peters, M.; Damen, L.; et al. Short-Term Hypoxia Dampens Inflammation in vivo via Enhanced Adenosine Release and Adenosine 2B Receptor Stimulation. EBioMedicine 2018, 33, 144–156.
- Kiers, H.D.; Scheffer, G.J.; van der Hoeven, J.G.; Eltzschig, H.K.; Pickkers, P.; Kox, M. Immunologic Consequences of Hypoxia during Critical Illness. Anesthesiology 2016, 125, 237–249.
- Hasko, G.; Antonioli, L.; Cronstein, B.N. Adenosine metabolism, immunity and joint health. Biochem. Pharmacol. 2018, 151, 307–313.
- Ferrari, D.; McNamee, E.N.; Idzko, M.; Gambari, R.; Eltzschig, H.K. Purinergic Signaling During Immune Cell Trafficking. Trends Immunol. 2016, 37, 399–411.
- Ferrari, D.; Bianchi, N.; Eltzschig, H.K.; Gambari, R. MicroRNAs Modulate the Purinergic Signaling Network. Trends Mol. Med. 2016, 22, 905–918.
- Idzko, M.; Ferrari, D.; Riegel, A.K.; Eltzschig, H.K. Extracellular nucleotide and nucleoside signaling in vascular and blood disease. Blood 2014, 124, 1029–1037.
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317.
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic signaling during inflammation. N. Engl. J. Med. 2012, 367, 2322–2333.
- Hasko, G.; Csoka, B.; Nemeth, Z.H.; Vizi, E.S.; Pacher, P. A(2B) adenosine receptors in immunity and inflammation. Trends Immunol. 2009, 30, 263–270.
- Koeppen, M.; Eckle, T.; Eltzschig, H.K. Selective deletion of the A1 adenosine receptor abolishes heart-rate slowing effects of intravascular adenosine in vivo. PLoS ONE 2009, 4, e6784.
- Sitkovsky, M.V.; Lukashev, D.; Apasov, S.; Kojima, H.; Koshiba, M.; Caldwell, C.; Ohta, A.; Thiel, M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu. Rev. Immunol. 2004, 22, 657–682.
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–920.
- Koscso, B.; Trepakov, A.; Csoka, B.; Nemeth, Z.H.; Pacher, P.; Eltzschig, H.K.; Hasko, G. Stimulation of A2B adenosine receptors protects against trauma-hemorrhagic shock-induced lung injury. Purinergic Signal. 2013, 9, 427–432.
- Nemeth, Z.H.; Lutz, C.S.; Csoka, B.; Deitch, E.A.; Leibovich, S.J.; Gause, W.C.; Tone, M.; Pacher, P.; Vizi, E.S.; Hasko, G. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J. Immunol. 2005, 175, 8260–8270.
- Cronstein, B.N.; Daguma, L.; Nichols, D.; Hutchison, A.J.; Williams, M. The adenosine/neutrophil paradox resolved: Human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J. Clin. Investig. 1990, 85, 1150–1157.
- Hasko, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770.
- Hasko, G.; Cronstein, B.N. Adenosine: An endogenous regulator of innate immunity. Trends Immunol. 2004, 25, 33–39.
- Loffler, M.; Morote-Garcia, J.C.; Eltzschig, S.A.; Coe, I.R.; Eltzschig, H.K. Physiological roles of vascular nucleoside transporters. Arter. Thromb. Vasc. Biol. 2007, 27, 1004–1013.
- Le, T.T.; Berg, N.K.; Harting, M.T.; Li, X.; Eltzschig, H.K.; Yuan, X. Purinergic Signaling in Pulmonary Inflammation. Front. Immunol. 2019, 10, 1633.
- Zhang, Y.; Dai, Y.; Wen, J.; Zhang, W.; Grenz, A.; Sun, H.; Tao, L.; Lu, G.; Alexander, D.C.; Milburn, M.V.; et al. Detrimental effects of adenosine signaling in sickle cell disease. Nat. Med. 2011, 17, 79–86.
- Van Linden, A.; Eltzschig, H.K. Role of pulmonary adenosine during hypoxia: Extracellular generation, signaling and metabolism by surface adenosine deaminase/CD26. Expert Opin. Biol. Ther. 2007, 7, 1437–1447.
- Eltzschig, H.K.; Faigle, M.; Knapp, S.; Karhausen, J.; Ibla, J.; Rosenberger, P.; Odegard, K.C.; Laussen, P.C.; Thompson, L.F.; Colgan, S.P. Endothelial catabolism of extracellular adenosine during hypoxia: The role of surface adenosine deaminase and CD26. Blood 2006, 108, 1602–1610.
- Eltzschig, H.K.; Eckle, T.; Mager, A.; Kuper, N.; Karcher, C.; Weissmuller, T.; Boengler, K.; Schulz, R.; Robson, S.C.; Colgan, S.P. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ. Res. 2006, 99, 1100–1108.
- Eltzschig, H.K.; Macmanus, C.F.; Colgan, S.P. Neutrophils as Sources of Extracellular Nucleotides: Functional Consequences at the Vascular Interface. Trends Cardiovasc. Med. 2008, 18, 103–107.
- Faigle, M.; Seessle, J.; Zug, S.; El Kasmi, K.C.; Eltzschig, H.K. ATP release from vascular endothelia occurs across Cx43 hemichannels and is attenuated during hypoxia. PLoS ONE 2008, 3, e2801.
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867.
- Ravichandran, K.S. Beginnings of a good apoptotic meal: The find-me and eat-me signaling pathways. Immunity 2011, 35, 445–455.
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286.
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367.
- Kaczmarek, E.; Koziak, K.; Sevigny, J.; Siegel, J.B.; Anrather, J.; Beaudoin, A.R.; Bach, F.H.; Robson, S.C. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J. Biol. Chem. 1996, 271, 33116–33122.
- Enjyoji, K.; Sevigny, J.; Lin, Y.; Frenette, P.S.; Christie, P.D.; Esch, J.S., 2nd; Imai, M.; Edelberg, J.M.; Rayburn, H.; Lech, M.; et al. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat. Med. 1999, 5, 1010–1017.
- Kohler, D.; Eckle, T.; Faigle, M.; Grenz, A.; Mittelbronn, M.; Laucher, S.; Hart, M.L.; Robson, S.C.; Muller, C.E.; Eltzschig, H.K. CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation 2007, 116, 1784–1794.
- Eckle, T.; Grenz, A.; Kohler, D.; Redel, A.; Falk, M.; Rolauffs, B.; Osswald, H.; Kehl, F.; Eltzschig, H.K. Systematic evaluation of a novel model for cardiac ischemic preconditioning in mice. Am. J. Physiol. Circ. Physiol. 2006, 291, H2533–H2540.
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136.
- Eltzschig, H.K.; Weissmuller, T.; Mager, A.; Eckle, T. Nucleotide metabolism and cell-cell interactions. Methods Mol. Biol. 2006, 341, 73–87.
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.A.; Enjyoji, K.; Robson, S.C.; Colgan, S.P. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: Role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 2003, 198, 783–796.
- Synnestvedt, K.; Furuta, G.T.; Comerford, K.M.; Louis, N.; Karhausen, J.; Eltzschig, H.K.; Hansen, K.R.; Thompson, L.F.; Colgan, S.P. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Investig. 2002, 110, 993–1002.
- Eckle, T.; Fullbier, L.; Wehrmann, M.; Khoury, J.; Mittelbronn, M.; Ibla, J.; Rosenberger, P.; Eltzschig, H.K. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J. Immunol. 2007, 178, 8127–8137.
- Eltzschig, H.K.; Thompson, L.F.; Karhausen, J.; Cotta, R.J.; Ibla, J.C.; Robson, S.C.; Colgan, S.P. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: Coordination by extracellular nucleotide metabolism. Blood 2004, 104, 3986–3992.
- Hart, M.L.; Gorzolla, I.C.; Schittenhelm, J.; Robson, S.C.; Eltzschig, H.K. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J. Immunol. 2010, 184, 4017–4024.
- Eltzschig, H.K.; Kohler, D.; Eckle, T.; Kong, T.; Robson, S.C.; Colgan, S.P. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood 2009, 113, 224–232.
- Colgan, S.P.; Eltzschig, H.K.; Eckle, T.; Thompson, L.F. Physiological roles for ecto-5’-nucleotidase (CD73). Purinergic Signal. 2006, 2, 351–360.
- Thompson, L.F.; Eltzschig, H.K.; Ibla, J.C.; Van De Wiele, C.J.; Resta, R.; Morote-Garcia, J.C.; Colgan, S.P. Crucial role for ecto-5’-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med. 2004, 200, 1395–1405.
- Eckle, T.; Krahn, T.; Grenz, A.; Kohler, D.; Mittelbronn, M.; Ledent, C.; Jacobson, M.A.; Osswald, H.; Thompson, L.F.; Unertl, K.; et al. Cardioprotection by ecto-5’-nucleotidase (CD73) and A2B adenosine receptors. Circulation 2007, 115, 1581–1590.
- Reichelt, M.E.; Willems, L.; Molina, J.G.; Sun, C.X.; Noble, J.C.; Ashton, K.J.; Schnermann, J.; Blackburn, M.R.; Headrick, J.P. Genetic deletion of the A1 adenosine receptor limits myocardial ischemic tolerance. Circ. Res. 2005, 96, 363–367.
- Auchampach, J.A.; Jin, X.; Moore, J.; Wan, T.C.; Kreckler, L.M.; Ge, Z.D.; Narayanan, J.; Whalley, E.; Kiesman, W.; Ticho, B.; et al. Comparison of three different A1 adenosine receptor antagonists on infarct size and multiple cycle ischemic preconditioning in anesthetized dogs. J. Pharmacol. Exp. Ther. 2004, 308, 846–856.
- Takano, H.; Bolli, R.; Black, R.G., Jr.; Kodani, E.; Tang, X.L.; Yang, Z.; Bhattacharya, S.; Auchampach, J.A. A(1) or A(3) adenosine receptors induce late preconditioning against infarction in conscious rabbits by different mechanisms. Circ. Res. 2001, 88, 520–528.
- Eckle, T.; Faigle, M.; Grenz, A.; Laucher, S.; Thompson, L.F.; Eltzschig, H.K. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood 2008, 111, 2024–2035.
- Revan, S.; Montesinos, M.C.; Naime, D.; Landau, S.; Cronstein, B.N. Adenosine A2 receptor occupancy regulates stimulated neutrophil function via activation of a serine/threonine protein phosphatase. J. Biol. Chem. 1996, 271, 17114–17118.
- Liu, H.; Zhang, Y.; Wu, H.; D’Alessandro, A.; Yegutkin, G.G.; Song, A.; Sun, K.; Li, J.; Cheng, N.Y.; Huang, A.; et al. Beneficial Role of Erythrocyte Adenosine A2B Receptor-Mediated AMP-Activated Protein Kinase Activation in High-Altitude Hypoxia. Circulation 2016, 134, 405–421.
- Song, A.; Zhang, Y.; Han, L.; Yegutkin, G.G.; Liu, H.; Sun, K.; D’Alessandro, A.; Li, J.; Karmouty-Quintana, H.; Iriyama, T.; et al. Erythrocytes retain hypoxic adenosine response for faster acclimatization upon re-ascent. Nat. Commun. 2017, 8, 14108.
- Hoegl, S.; Brodsky, K.S.; Blackburn, M.R.; Karmouty-Quintana, H.; Zwissler, B.; Eltzschig, H.K. Alveolar Epithelial A2B Adenosine Receptors in Pulmonary Protection during Acute Lung Injury. J. Immunol. 2015, 195, 1815–1824.
- Eckle, T.; Hughes, K.; Ehrentraut, H.; Brodsky, K.S.; Rosenberger, P.; Choi, D.S.; Ravid, K.; Weng, T.; Xia, Y.; Blackburn, M.R.; et al. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury. FASEB J. 2013, 27, 3078–3089.
- Hesse, J.; Groterath, W.; Owenier, C.; Steinhausen, J.; Ding, Z.; Steckel, B.; Czekelius, C.; Alter, C.; Marzoq, A.; Schrader, J. Normoxic induction of HIF-1alpha by adenosine-A2B R signaling in epicardial stromal cells formed after myocardial infarction. FASEB J. 2021, 35, e21517.
- Aherne, C.M.; Saeedi, B.; Collins, C.B.; Masterson, J.C.; McNamee, E.N.; Perrenoud, L.; Rapp, C.R.; Curtis, V.F.; Bayless, A.; Fletcher, A.; et al. Epithelial-specific A2B adenosine receptor signaling protects the colonic epithelial barrier during acute colitis. Mucosal. Immunol. 2015, 8, 699.
- Ehrentraut, H.; Westrich, J.A.; Eltzschig, H.K.; Clambey, E.T. Adora2b Adenosine Receptor Engagement Enhances Regulatory T Cell Abundance during Endotoxin-Induced Pulmonary Inflammation. PLoS ONE 2012, 7, e32416.
- Lu, B.; Rajakumar, S.V.; Robson, S.C.; Lee, E.K.; Crikis, S.; d’Apice, A.J.; Cowan, P.J.; Dwyer, K.M. The impact of purinergic signaling on renal ischemia-reperfusion injury. Transplantation 2008, 86, 1707–1712.
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265.
- Alter, C.; Ding, Z.; Flogel, U.; Scheller, J.; Schrader, J. A2bR-dependent signaling alters immune cell composition and enhances IL-6 formation in the ischemic heart. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H190–H200.
- Kong, T.; Westerman, K.A.; Faigle, M.; Eltzschig, H.K.; Colgan, S.P. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J. 2006, 20, 2242–2250.
- Eckle, T.; Kewley, E.M.; Brodsky, K.S.; Tak, E.; Bonney, S.; Gobel, M.; Anderson, D.; Glover, L.E.; Riegel, A.K.; Colgan, S.P.; et al. Identification of hypoxia-inducible factor HIF-1A as transcriptional regulator of the A2B adenosine receptor during acute lung injury. J. Immunol. 2014, 192, 1249–1256.
- Ahmad, A.; Ahmad, S.; Glover, L.; Miller, S.M.; Shannon, J.M.; Guo, X.; Franklin, W.A.; Bridges, J.P.; Schaack, J.B.; Colgan, S.P.; et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10684–10689.
- Yang, Z.; Day, Y.J.; Toufektsian, M.C.; Ramos, S.I.; Marshall, M.; Wang, X.Q.; French, B.A.; Linden, J. Infarct-sparing effect of A2A-adenosine receptor activation is due primarily to its action on lymphocytes. Circulation 2005, 111, 2190–2197.
- Yang, Z.; Day, Y.J.; Toufektsian, M.C.; Xu, Y.; Ramos, S.I.; Marshall, M.A.; French, B.A.; Linden, J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 2006, 114, 2056–2064.
- Maas, J.E.; Wan, T.C.; Figler, R.A.; Gross, G.J.; Auchampach, J.A. Evidence that the acute phase of ischemic preconditioning does not require signaling by the A 2B adenosine receptor. J. Mol. Cell. Cardiol. 2010, 49, 886–893.
- Seo, S.W.; Koeppen, M.; Bonney, S.; Gobel, M.; Thayer, M.; Harter, P.N.; Ravid, K.; Eltzschig, H.K.; Mittelbronn, M.; Walker, L.; et al. Differential Tissue-Specific Function of Adora2b in Cardioprotection. J. Immunol. 2015, 195, 1732–1743.
- Koeppen, M.; Harter, P.N.; Bonney, S.; Bonney, M.; Reithel, S.; Zachskorn, C.; Mittelbronn, M.; Eckle, T. Adora2b Signaling on Bone Marrow Derived Cells Dampens Myocardial Ischemia-Reperfusion Injury. Anesthesiology 2012, 116, 1245–1257.
- Bonney, S.; Kominsky, D.; Brodsky, K.; Eltzschig, H.; Walker, L.; Eckle, T. Cardiac Per2 functions as novel link between fatty acid metabolism and myocardial inflammation during ischemia and reperfusion injury of the heart. PLoS ONE 2013, 8, e71493.
- Ruan, W.; Yuan, X.; Eltzschig, H. Circadian Mechanisms in Medicine. N. Engl. J. Med. 2021, 384, e76.
- Ruan, W.; Yuan, X.; Eltzschig, H.K. Circadian rhythm as a therapeutic target. Nat. Rev. Drug Discov. 2021, 20, 287–307.
- Keller, M.; Mirakaj, V.; Koeppen, M.; Rosenberger, P. Neuronal guidance proteins in cardiovascular inflammation. Basic Res. Cardiol. 2021, 116, 6.
- Mirakaj, V.; Gatidou, D.; Potzsch, C.; Konig, K.; Rosenberger, P. Netrin-1 signaling dampens inflammatory peritonitis. J. Immunol. 2011, 186, 549–555.
- Mirakaj, V.; Thix, C.A.; Laucher, S.; Mielke, C.; Morote-Garcia, J.C.; Schmit, M.A.; Henes, J.; Unertl, K.E.; Kohler, D.; Rosenberger, P. Netrin-1 dampens pulmonary inflammation during acute lung injury. Am. J. Respir. Crit. Care Med. 2010, 181, 815–824.
- Serafini, T.; Colamarino, S.A.; Leonardo, E.D.; Wang, H.; Beddington, R.; Skarnes, W.C.; Tessier-Lavigne, M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 1996, 87, 1001–1014.
- Serafini, T.; Kennedy, T.E.; Galko, M.J.; Mirzayan, C.; Jessell, T.M.; Tessier-Lavigne, M. The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell 1994, 78, 409–424.
- Varadarajan, S.G.; Kong, J.H.; Phan, K.D.; Kao, T.J.; Panaitof, S.C.; Cardin, J.; Eltzschig, H.; Kania, A.; Novitch, B.G.; Butler, S.J. Netrin1 Produced by Neural Progenitors, Not Floor Plate Cells, Is Required for Axon Guidance in the Spinal Cord. Neuron 2017, 94, 790–799.e3.
- Gao, R.; Peng, X.; Perry, C.; Sun, H.; Ntokou, A.; Ryu, C.; Gomez, J.L.; Reeves, B.C.; Walia, A.; Kaminski, N.; et al. Macrophage-derived netrin-1 drives adrenergic nerve-associated lung fibrosis. J. Clin. Investig. 2021, 131, e136542.
- Moore, S.W.; Tessier-Lavigne, M.; Kennedy, T.E. Netrins and their receptors. Adv. Exp. Med. Biol. 2007, 621, 17–31.
- Hadi, T.; Boytard, L.; Silvestro, M.; Alebrahim, D.; Jacob, S.; Feinstein, J.; Barone, K.; Spiro, W.; Hutchison, S.; Simon, R.; et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nat. Commun. 2018, 9, 5022.
- Mirakaj, V.; Rosenberger, P. Immunomodulatory Functions of Neuronal Guidance Proteins. Trends Immunol. 2017, 38, 444–456.
- Corset, V.; Nguyen-Ba-Charvet, K.T.; Forcet, C.; Moyse, E.; Chedotal, A.; Mehlen, P. Netrin-1-mediated axon outgrowth and cAMP production requires interaction with adenosine A2b receptor. Nature 2000, 407, 747–750.
- Chen, Z.; Chen, Y.; Zhou, J.; Li, Y.; Gong, C.; Wang, X. Netrin-1 reduces lung ischemia-reperfusion injury by increasing the proportion of regulatory T cells. J. Int. Med. Res. 2020, 48, 300060520926415.
- He, J.; Zhao, Y.; Deng, W.; Wang, D.X. Netrin-1 promotes epithelial sodium channel-mediated alveolar fluid clearance via activation of the adenosine 2B receptor in lipopolysaccharide-induced acute lung injury. Respiration 2014, 87, 394–407.
- Aherne, C.M.; Collins, C.B.; Eltzschig, H.K. Netrin-1 guides inflammatory cell migration to control mucosal immune responses during intestinal inflammation. Tissue Barriers 2013, 1, e24957.
- Aherne, C.M.; Collins, C.B.; Masterson, J.C.; Tizzano, M.; Boyle, T.A.; Westrich, J.A.; Parnes, J.A.; Furuta, G.T.; Rivera-Nieves, J.; Eltzschig, H.K. Neuronal guidance molecule netrin-1 attenuates inflammatory cell trafficking during acute experimental colitis. Gut 2012, 61, 695–705.
- Tak, E.; Ridyard, D.; Badulak, A.; Giebler, A.; Shabeka, U.; Werner, T.; Clambey, E.; Moldovan, R.; Zimmerman, M.A.; Eltzschig, H.K.; et al. Protective role for netrin-1 during diabetic nephropathy. J. Mol. Med. 2013, 91, 1071–1080.
- Zhang, Y.; Chen, P.; Di, G.; Qi, X.; Zhou, Q.; Gao, H. Netrin-1 promotes diabetic corneal wound healing through molecular mechanisms mediated via the adenosine 2B receptor. Sci. Rep. 2018, 8, 5994.
- Li, J.; Conrad, C.; Mills, T.W.; Berg, N.K.; Kim, B.; Ruan, W.; Lee, J.W.; Zhang, X.; Yuan, X.; Eltzschig, H.K. PMN-derived netrin-1 attenuates cardiac ischemia-reperfusion injury via myeloid ADORA2B signaling. J. Exp. Med. 2021, 218, e20210008.
- Stein, E.; Zou, Y.; Poo, M.; Tessier-Lavigne, M. Binding of DCC by netrin-1 to mediate axon guidance independent of adenosine A2B receptor activation. Science 2001, 291, 1976–1982.
- Rosenberger, P.; Schwab, J.M.; Mirakaj, V.; Masekowsky, E.; Mager, A.; Morote-Garcia, J.C.; Unertl, K.; Eltzschig, H.K. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat. Immunol. 2009, 10, 195–202.
- Ramkhelawon, B.; Yang, Y.; van Gils, J.M.; Hewing, B.; Rayner, K.J.; Parathath, S.; Guo, L.; Oldebeken, S.; Feig, J.L.; Fisher, E.A.; et al. Hypoxia induces netrin-1 and Unc5b in atherosclerotic plaques: Mechanism for macrophage retention and survival. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1180–1188.
- Griffiths, M.; Beaumont, N.; Yao, S.Y.; Sundaram, M.; Boumah, C.E.; Davies, A.; Kwong, F.Y.; Coe, I.; Cass, C.E.; Young, J.D.; et al. Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat. Med. 1997, 3, 89–93.
- Picano, E.; Trivieri, M.G. Pharmacologic stress echocardiography in the assessment of coronary artery disease. Curr. Opin. Cardiol. 1999, 14, 464–470.
- Sicari, R.; Cortigiani, L.; Bigi, R.; Landi, P.; Raciti, M.; Picano, E. Prognostic value of pharmacological stress echocardiography is affected by concomitant antiischemic therapy at the time of testing. Circulation 2004, 109, 2428–2431.
- Wang, W.; Chen, N.Y.; Ren, D.; Davies, J.; Philip, K.; Eltzschig, H.K.; Blackburn, M.R.; Akkanti, B.; Karmouty-Quintana, H.; Weng, T. Enhancing Extracellular Adenosine Levels Restores Barrier Function in Acute Lung Injury Through Expression of Focal Adhesion Proteins. Front. Mol. Biosci. 2021, 8, 636678.
- Aherne, C.M.; Collins, C.B.; Rapp, C.R.; Olli, K.E.; Perrenoud, L.; Jedlicka, P.; Bowser, J.L.; Mills, T.W.; Karmouty-Quintana, H.; Blackburn, M.R.; et al. Coordination of ENT2-dependent adenosine transport and signaling dampens mucosal inflammation. JCI Insight 2018, 3, e121521.
- Morote-Garcia, J.C.; Kohler, D.; Roth, J.M.; Mirakaj, V.; Eldh, T.; Eltzschig, H.K.; Rosenberger, P. Repression of the equilibrative nucleoside transporters dampens inflammatory lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 296–305.
- Rose, J.B.; Naydenova, Z.; Bang, A.; Ramadan, A.; Klawitter, J.; Schram, K.; Sweeney, G.; Grenz, A.; Eltzschig, H.; Hammond, J.; et al. Absence of equilibrative nucleoside transporter 1 in ENT1 knockout mice leads to altered nucleoside levels following hypoxic challenge. Life Sci. 2011, 89, 621–630.
- Kitakaze, M.; Minamino, T.; Node, K.; Takashima, S.; Funaya, H.; Kuzuya, T.; Hori, M. Adenosine and cardioprotection in the diseased heart. Jpn. Circ. J. 1999, 63, 231–243.
- Miura, T.; Ogawa, T.; Iwamoto, T.; Shimamoto, K.; Iimura, O. Dipyridamole potentiates the myocardial infarct size-limiting effect of ischemic preconditioning. Circulation 1992, 86, 979–985.
- Morote-Garcia, J.C.; Rosenberger, P.; Kuhlicke, J.; Eltzschig, H.K. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood 2008, 111, 5571–5580.
- Peart, J.N.; Gross, G.J. Cardioprotection following adenosine kinase inhibition in rat hearts. Basic Res. Cardiol. 2005, 100, 328–336.