Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Pulmonary fibrosis (PF) is a feared outcome of many pulmonary diseases which results in a reduction in lung compliance and capacity. The development of PF is relatively rare, but it can occur secondary to viral pneumonia, especially COVID-19 infection. While COVID-19 infection and its complications are still under investigation, researchers can look at a similar outbreak in the past to gain better insight as to the expected long-term outcomes of COVID-19 patient lung function.
- pulmonary fibrosis
- COVID-19
- TGF-β1
- prevention
1. Introduction
Pulmonary fibrosis (PF) is a feared outcome of many pulmonary diseases as the progression of PF results in a reduction in lung compliance and capacity as well as an increase in alveolar wall thickness. These changes combine to result in poor lung function and labored respiration in afflicted patients. The mechanisms underlying PF are complex and currently not completely characterized due to its many etiologies. The development of PF is relatively rare, but it can occur secondary to viral pneumonia, especially COVID-19 [1]. With the massive scale of the COVID-19 pandemic, researchers expect there should more instances of PF due to COVID-19 infection.
While COVID-19 infection and its complications are still under investigation, researchers can look at a similar outbreak in the past to gain better insight as to the expected long-term outcomes of COVID-19 patient lung function. When compared to a similar infection, severe acute respiratory syndrome (SARS), which first appeared in China in 2002, some patients experienced a decline in respiratory function after the infection. A study looking at the pulmonary function of 97 patients who survived SARS in a one-year follow-up found that 28% had chest X-ray abnormalities. The pulmonary function testing shows that 4% of patients had a decreased forced vital capacity (FVC), 5% had a decrease in total lung capacity (TLC), and 24% had a decreased diffusion capacity for carbon monoxide (DLCO). A total of 32% of the patients that were admitted to the intensive care unit reported having a worse quality of life [2]. Overall, studies looking at SARS survivors and long-term health outcomes show 28% to 53% percent having decreased lung function similar to that of symptoms of pulmonary fibrosis [2][3][4]. This shows that post-COVID-19 PF is a significant concern.
2. Pathophysiology of Pulmonary Fibrosis
The pathophysiology of PF is an area of ongoing research. This disease state can result from many etiologies which may result in different primary mechanisms that drive the shared fibrotic outcome. Patients who develop acute respiratory distress syndrome (ARDS) [5] may experience in an excessive proliferation of fibrotic factors leading to pulmonary fibrosis. Other patients may develop fibrosis from environmental insults, such as chest irradiation or chronic occupational exposure to irritants such as asbestos or silica. Rarely, patients may develop IPF, which is a severe, progressive fibrotic disease of pulmonary tissue with an unknown insult (Figure 1).
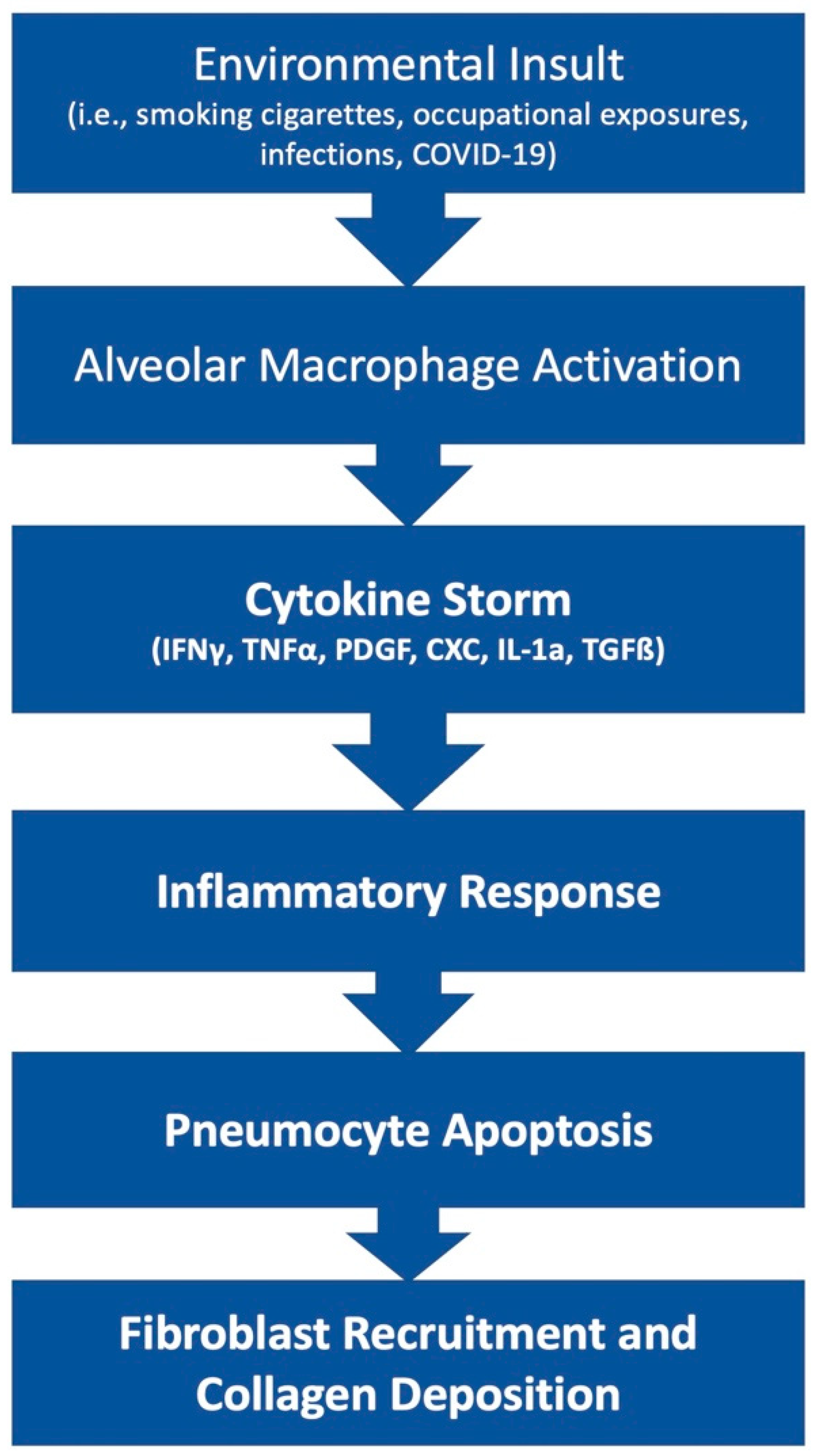
Figure 1. Simplified diagram of the development of fibrosis.
The lung tissue’s response to irritants or disease states can either result in resolution and return to normal function or the development of fibrosis that leads to a permanent reduction in total lung capacity. Here, researchers explore the possible interactions that favor the development of fibrosis. Researchers focus on Alveolar epithelial cells (AECs), various cytokines, transforming growth factor (TGF)-β, myofibroblasts, and environmental factors as they relate to the development of fibrosis post-COVID-19 infection.
AECs are an important group of cells within the parenchyma of the lungs that are responsible for many aspects of inflammatory regulation. AECs have two subtypes, AEC type I (AEC1) and AEC type II (AEC2), with both different roles and functions. AEC1 forms a simple squamous layer that normally surrounds the alveolar airspace and is involved primarily in gas exchange. The role of AEC2 cells is more complex. AEC2 cells can produce surfactant, a protein–lipid secretion that lowers the surface tension forces acting on the alveolus. AEC2 cells also serve as the predominant epithelial progenitor cells that are capable of differentiation into AEC1 cells. Loss of function or reduced AEC cell counts can lead to improper repair of the lung parenchyma, which can then lead to fibrosis [6].
The development of PF may be driven by microenvironmental insults to AECs. These microenvironmental insults can be from a variety of intrinsic (genetic, age, gender) or extrinsic factors such as infection (COVID-19), cigarette smoke, or occupational exposure. After the initial insult, the AEC2 cells may stimulate an immune response to clear or contain the microenvironmental insult. Optimally, once the insults are cleared, the inflammatory response subsides and the remaining AEC2s proliferate to repair damage to the tissue, ultimately leading to resolution. However, if the insults lead to the dysfunction of the AEC2, the cells will stimulate the recruitment of myofibroblasts. Myofibroblasts will lead to the activation of collagen synthesis, apoptosis of AEC2 cells, and ultimately, PF [7]. This dysregulation of the healing response and dysfunction of AEC2 cells is associated with long-lasting or recurring insults. The longer the insults take hold within the lung, the less likely a complete resolution will take place. Minimizing the offending insult quickly will play a key role in the prevention of fibrosis.
In addition to the AECs within the pulmonary tissue, many cytokines are involved in the upregulation of the inflammation that ultimately leads to fibrosis. The role of inflammatory cytokines in the progression of the PF is complex. The major stimulators include TGF-β. The role of TGF-β in PF is widespread, with many different mediators. A major cellular source of TGF-β is alveolar macrophages, bronchial epithelium, and hyperplastic AEC2 in response to inflammation and alveolar damage [8]. A primary role of TGF-β is to modulate the extracellular matrix (ECM) deposition through the activation of myofibroblasts. TGF-β seems to activate P120-catenin, an adhesion protein, to increase the fibroblastic foci and primary fibroblast in Bleomycin-challenged mice [9]. Bleomycin is a drug that is known to induce pulmonary fibrosis. Fibroblastic foci are a characteristic of idiopathic PF which indicate an area of lung destruction. TGF-β1, a class of TGF-β, regulates cell recruitment to sites of injury, induces fibroblast differentiation to myofibroblast, stimulates ECM production by myofibroblast, and inhibits the ECM degradation by matrix metalloproteinase [10]. There are also increased alveolar secretions of fibroblast growth factor (FGF)-2 in response to an increase in TGF-β1 activity through the activation of the extracellular-signal-regulated kinase (ERK) pathway. TGF-β1 response through the ERK kinase pathway and treatment with ERK1/2 inhibitors seems to attenuate the fibrotic differentiation [11]. Finally, TGF-β is regulated through the WNT/B-catenin pathway, and WNT10 overexpression has been shown to increase exacerbation and poor prognosis among IPF patients [12]. Given the many pro-fibrotic effects of TGF-β in the lung, targeting of TGF-β is theorized to be a desirable target for therapy in hope of preventing the progression of fibrosis.
Interestingly, there is a shift in the local cytokine profile in PF patients in addition to increased TGF-β signaling. A shift from a TH2 inflammatory profile to a TH1 inflammatory profile seems to decrease the progression of fibrosis [13]. The TH1 inflammatory profile includes the interferon-gamma (IFN)-γ and Interleukin (IL)-12; both attenuate the fibrosis whereas the TH2 inflammatory profile includes the IL-4, IL-5, and IL-13, which have been linked to fibrogenesis [14]. Other cytokines such as Tumor necrosis factor (TNF)-α, Platelet-derived growth factor (PDGF), CXC Chemokines, IL-1a, and TGF-β have been linked to the development of IPF [15]. The implication of the cytokines in IPF has prompted more investigation of different immunotherapies.
In addition to the cytokine response, the lung response to insult or injury includes the promotion of myofibroblasts. Myofibroblasts are differentiated from fibroblasts and then quickly recruited to the lung to facilitate the repair processes. Myofibroblasts migrate and secrete ECM within the area of damage, which provides a fundamental scaffolding for the repair process. The deposition of ECM can also serve as protective scar tissue by walling off the area of damage and preventing the spread of inflammation to the healthy areas. However, an imbalance in the ECM deposition leads to the formation of fibrosis. The stiffening of the ECM has been associated with the progression of IPF [16]. The regulation of myofibroblast activation and proliferation is a complex procedure and is currently under investigation. It is known that there are many factors facilitating its differentiation, including TGF- β1 through the microRNA-133a [17], or through Src family kinase [18]. Attenuating the differentiation of myofibroblast and limiting the collagen deposition may be a good strategy to slow the progression of pulmonary fibrosis.
Among others, oxidative stress is also implicated in the development of fibrosis. Reactive oxygen species (ROS) can be formed in excess from the innate inflammatory processes, such as in neutrophil granules, or environmental exposure. The lungs are especially vulnerable to oxidative stress, as the level of O2 is much higher than the rest of the body. High-level ROS is theorized to induce epithelial apoptosis, increase secretion of profibrotic cytokine, and increase differentiation of fibroblast to myofibroblast [19]. Of note, severe COVID-19 patients often need the ventilators to assist with breathing, which can increase the risk of oxidative stress in these patients.
COVID-19 patients also have an increased risk of pulmonary embolism (PE), further exacerbating the already decreased lung function. Patients with COVID-19 have an overall 16.5% incidence rate of developing a PE [20]. The hypercoagulability state that these patients are in predisposed them to developing chronic PE, which could also exacerbate the progression of PF.
This entry is adapted from the peer-reviewed paper 10.3390/cells11162489
References
- Gentile, F.; Aimo, A.; Forfori, F.; Catapano, G.; Clemente, A.; Cademartiri, F.; Emdin, M.; Giannoni, A. COVID-19 and risk of pulmonary fibrosis: The importance of planning ahead. Eur. J. Prev. Cardiol. 2020, 27, 1442–1446.
- Hui, D.S.; Wong, K.T.; Ko, F.W.; Tam, L.S.; Chan, D.P.; Woo, J.; Sung, J.J. The 1-year impact of severe acute respiratory syndrome on pulmonary function, exercise capacity, and quality of life in a cohort of survivors. Chest 2005, 128, 2247–2261.
- Ngai, J.C.; Ko, F.W.; Ng, S.S.; To, K.W.; Tong, M.; Hui, D.S. The long-term impact of severe acute respiratory syndrome on pulmonary function, exercise capacity and health status. Respirology 2010, 15, 543–550.
- Chang, Y.C.; Yu, C.J.; Chang, S.C.; Galvin, J.R.; Liu, H.M.; Hsiao, C.H.; Kuo, P.H.; Chen, K.Y.; Franks, T.J.; Huang, K.M.; et al. Pulmonary sequelae in convalescent patients after severe acute respiratory syndrome: Evaluation with thin-section CT. Radiology 2005, 236, 1067–1075.
- Tomashefski, J.F., Jr. Pulmonary pathology of acute respiratory distress syndrome. Clin. Chest Med. 2000, 21, 435–466.
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int. J. Mol. Sci. 2020, 21, 2269.
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099.
- Yue, X.; Shan, B.; Lasky, J.A. TGF-β: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6, 67–77.
- Zhang, Y.; Jiao, H.; Wu, Y.; Sun, X. P120-catenin regulates pulmonary fibrosis and TGF-β induced lung fibroblast differentiation. Life Sci. 2019, 230, 35–44.
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69.
- Xiao, L.; Du, Y.; Shen, Y.; He, Y.; Zhao, H.; Li, Z. TGF-beta 1 induced fibroblast proliferation is mediated by the FGF-2/ERK pathway. Front. Biosci. 2012, 17, 2667–2674.
- Oda, K.; Yatera, K.; Izumi, H.; Ishimoto, H.; Yamada, S.; Nakao, H.; Hanaka, T.; Ogoshi, T.; Noguchi, S.; Mukae, H. Profibrotic role of WNT10A via TGF-ß signaling in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 39.
- Kikuchi, N.; Ishii, Y.; Morishima, Y.; Yageta, Y.; Haraguchi, N.; Itoh, K.; Yamamoto, M.; Hizawa, N. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir. Res. 2010, 11, 31.
- Kolahian, S.; Fernandez, I.E.; Eickelberg, O.; Hartl, D. Immune Mechanisms in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 309–322.
- Agostini, C.; Gurrieri, C. Chemokine/cytokine cocktail in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 357–363.
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 180033.
- Wei, P.; Xie, Y.; Abel, P.W.; Huang, Y.; Ma, Q.; Li, L.; Hao, J.; Wolff, D.W.; Wei, T.; Tu, Y. Transforming growth factor (TGF)-β1-induced miR-133a inhibits myofibroblast differentiation and pulmonary fibrosis. Cell Death Dis. 2019, 10, 670.
- Li, H.; Zhao, C.; Tian, Y.; Lu, J.; Zhang, G.; Liang, S.; Chen, D.; Liu, X.; Kuang, W.; Zhu, M. Src family kinases and pulmonary fibrosis: A review. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 127, 110183.
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1028–1040.
- Suh, Y.J.; Hong, H.; Ohana, M.; Bompard, F.; Revel, M.P.; Valle, C.; Gervaise, A.; Poissy, J.; Susen, S.; Hékimian, G.; et al. Pulmonary Embolism and Deep Vein Thrombosis in COVID-19: A Systematic Review and Meta-Analysis. Radiology 2021, 298, E70–E80.
This entry is offline, you can click here to edit this entry!