Facing greenhouse effects and the rapid exhaustion of fossil fuel, CO2 electrochemical reduction presents a promising method of environmental protection and energy transformation. Low onset potential, large current density, high faradaic efficiency (FE), and long-time stability are required for industrial production, due to economic costs and energy consumption.
1. Introduction
In recent years, the increasing usage of fossil fuels has not only accelerated the exhaustion of limited natural energy, but has also led to enormous CO
2 emissions, resulting in energy shortages and greenhouse effects. A great deal of research has been undertaken to solve these problems. Among many suggestions, electrochemical CO
2 reduction reaction (CO
2RR) appears a promising way to produce value-added chemicals and simultaneously decrease CO
2 concentration in the atmosphere [
1,
2]. Powered by intermittent renewable electricity, and using water as the proton donor, CO
2RR converts CO
2 molecules into a wide range of reduced carbon compounds, including carbon monoxide (CO), methane (CH
4), formic acid (HCOOH), methanol (CH
3OH), ethylene (C
2H
4), ethanol (C
2H
5OH), and acetate (CH
3COOH) [
3]. The collected products are either directly used as fuels or further converted into value-added chemicals [
4,
5]. With these advantages, CO
2RR has become an important research area and many scientists have tried to put the associated ideas into practice.
The low current density, low faradaic efficiency (FE), and high overpotential occurring with CO
2RR remain a challenge for product generation. The disadvantages of the stable C=O bond (about 750 kJ mol
−1) [
6], and the complex pathway of proton-coupled electron transfer, contribute to the chemical sluggishness of CO
2RR [
7]. The similar range of redox potentials for different CO
2RR products and the hydrogen evolution reaction (HER) contribute to the poor Faradaic efficiency (FE) of specific products [
8]. Although many electrochemical catalysts have been designed to enhance the activity and selectivity, and good catalytic performance was obtained, another issue of catalytic stability was revealed in further application. Stability should be maintained for at least 4000 h for industrial application, and more than 20,000 h are required to make production appealing economically [
9,
10]. However, various factors in electroreduction, including surface reconstruction, flooding behavior, etc., caused unsatisfactory stability only lasting for tens of hours, far from industrial requirements [
11,
12]. Thus, research was carried out targeting large current density, high FE, low overpotential, and long stability.
Since Hori’s first work in 1985 [
13], many efforts in fields of mechanism exploration and catalyst synthesis have been made to disclose the reaction process and to overcome the obstacles mentioned above [
14,
15,
16,
17,
18,
19,
20,
21,
22]. Methods for catalyst synthesis including morphology [
23], facet engineering [
24,
25], metal doping [
26,
27], and surface modification [
28,
29] could modulate the binding strength between the intermediates and active centers, thus favoring reaction rates and rearranging product distribution. Copper is one of the few metals that can reduce CO
2 to hydrocarbons and alcohols with decent efficiency, and copper-based catalysts have received much attention [
30,
31]. The uniqueness of Cu as a CO
2RR electrocatalyst is explained by the fact that it is the only metal that has negative adsorption energy for *CO and positive adsorption energy for *H [
2]. Furthermore, the moderate binding of *CO to Cu provides a balance between activation of CO
2 and hydrogenation of *CO, which is the key step towards hydrocarbons. In view of the unique advantage of Cu, we will focus on Cu-based catalysts and strategies to improve their catalytic performance.
In addition to catalyst design, other improvements to the catalytic system have been presented in recent years. Gas diffusion electrodes (GDE) were widely adopted to feed reactant gas directly to the interface between the catalyst and electrolyte, enhancing the CO
2 concentration on the catalysts’ surface and promoting the current density. Other methods including changing the CO
2 concentration and electrolyte engineering were also reported to enhance catalytic performance [
32,
33].
2. Electroreduction Pathways
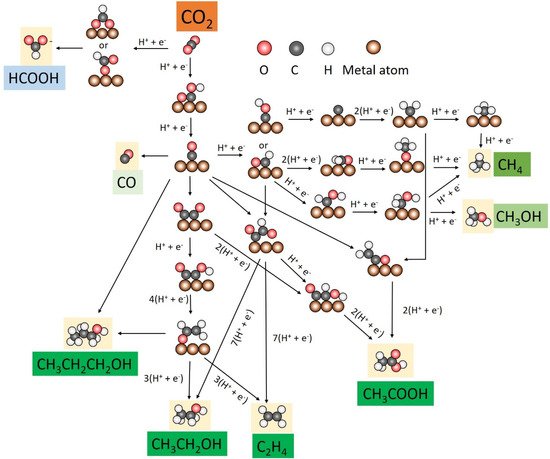
CO2RR activity is carried out through multiple proton and electron transfer steps involving many possible intermediates. The catalytic performance can be tuned and determined largely by key reaction intermediates. Advances in operando spectroscopy and computational techniques provide significant scope for exploring the evolution of surface-bound species and rationalizing a pathway to the desired product. The reaction pathways of CO2 reduction with key intermediates on copper-based catalysts are summarized (Figure 1).
Figure 1. CO2RR pathways towards C1 and C2+ products on metal-based catalysts.
The reaction pathways for the formation of C
1 products are relatively simple. C
1 products from CO
2RR include HCOOH, CO, CH
3OH, and CH
4. Generally, the formate pathway, which involves the *OCHO intermediate, is considered a dead-end, and the *OCHO is bound to the catalyst surface via one or two oxygen atoms [
19,
35]. For CO, *COOH is first generated with carbon atom binding to the catalyst surface, followed by *CO formation through dehydrogenation of *COOH, which is finally desorbed from the catalyst to release CO [
36]. *CO is the initial key intermediate for further reduction, and the selectivity is determined by the binding energy of *CO. If the binding energy is too weak, the *CO will be desorbed from the catalyst surface to generate CO. If the catalyst binds *CO very strongly, the catalytically active sites are poisoned and the HER becomes dominant. Only the catalysts with the moderate binding energy of *CO can produce muti-electron reduction products [
37]. *CO is further reduced to CH
4 through the *CHO pathway on these catalysts, forming *CHOH and then *CH
2OH and finally branching into two routes (CH
3OH and CH
4) [
38]. It has also been observed that *C is formed by dehydration of *COH and constitutes another pathway to generate CH
4 [
39]. Furthermore, *CH
3O formed by protonation of *CHO is also an important intermediate for CH
4 generation [
40,
41].
The C
2+ pathways are much more complex and many controversies still exist. The electroreduction pathway towards the main products includes C
2H
4, C
2H
5OH, CH
3COOH, and n-C
3H
7OH. In the formation of the multi-carbon compounds, C–C coupling is the most important step and starts between different C-containing intermediates. *CO dimerization is the most widely accepted step, having been evidenced by theoretical calculation and in situ spectroscopic observation [
42,
43]. Then, the *CO dimer is further reduced to *OCHCH
2, which serves as the key intermediate in determining the selectivity of ethylene and ethanol [
44,
45]. It is proposed that the *CO dimer follows another pathway to produce CH
3COOH via *COCHOH intermediate [
3]. Additionally, some studies suggest the possibility of coupling between *CHO and *CO, especially at higher overpotentials. After coupling, the intermediate *COCHO is further reduced to ethanol, ethylene, and acetate [
18]. The coupling occurs between *CO and *CH
2 via *CO insertion, which produces acetate through multiple proton and electron transfer steps [
34,
46]. Although it was suggested that
n-propanol forms by C–C coupling between CO and C
2H
4 precursors [
47], the elementary steps for C
3 production have not yet been well proposed.
3. Advances in CO2 Electroreduction
The recent research into CO2 electroreduction focuses on reducing overpotential, enhancing the FE of the target product, enlarging current density, and maintaining stability. Different synthesis methods of the catalysts and catalysis engineering have been introduced to promote catalytic performance.
3.1. Low Overpotential
Overpotential is defined as the additional potential (above the thermodynamic requirement) to drive a reaction at a specific current density [
48]. It can be simulated by theoretical calculations such as density functional theory (DFT). The adsorption energies of the evolving intermediates were calculated to build up the thermodynamic energy diagram for CO
2RR, and the overpotential was attributed to the most unfavorable step in the electroreduction pathway [
27,
40], which was called the rate-determining step (RDS). Although some methods of calculation correction have been developed to assess the kinetic barrier [
49,
50], the difference in adsorption energy of intermediates was found to influence the overpotential, and other effects such as local CO concentration and charge transfer resistance also played a vital role. Recent research on reducing the onset potential for CO
2RR products is shown in
Table 1. The potential for the target product is quite different. Low overpotential is required for CO and formate, while high overpotential is needed for deep electroreduction products of methane, ethanol, and ethylene.
Table 1. Recent research on onset potential for CO2RR products (* The onset potential is not clearly determined and this value is the lowest applied bias with decent FE).
| Major Product |
Catalyst |
Onset Potential
(V vs. RHE) |
Reference |
| CO |
Cu-N2/CN |
−0.33 |
[51] |
| FeN5 |
−0.2 * |
[52] |
| Ni–N3S |
−0.17 |
[53] |
| Fe3+-N-C |
−0.2 |
[54] |
| Co–N–Ni/NPCNSs |
−0.2 |
[55] |
| CoPc©Fe-N-C |
−0.13 |
[56] |
| Formate |
single-atom Snδ+ on N-doped graphene |
−0.18 |
[57] |
| BiN4/C |
−0.51 |
[58] |
| Methane |
AuAgPtPdCu |
−0.3 * |
[59] |
| Ethylene |
Organosuperbases modified Cu-NC |
−0.43 |
[60] |
| F-Cu |
~−0.2 |
[61] |
| Ethanol |
Cun (n = 3 and 4) cluster |
−0.3~−0.4 |
[62] |
| Au/Cu |
−0.7 * |
[63] |
| FeTPP[Cl]/Cu |
−0.42 |
[64] |
| Acetate |
Cu–Cu2O/Cu |
~−0.2 * |
[65] |
3.1.1. Two-Electron Electroreduction Products
Generally, two-electron electroreduction products with a relatively simple process require lower overpotential, compared with other reduction products such as methane and ethylene. Since the properties of active metal centers in SACS can be finely tuned through changing the near-range coordination environment and long-range interactions, SACs could effectively lower the overpotential of CO2RR products. Considering the promotion effect of unsaturated coordination on catalytic activity, Zheng et al. fabricated coordinatively unsaturated single-atom with nitrogen sites anchored on graphene (Cu-N2/CN) [51]. Aberration corrected high-angle annular dark field scanning transmission electron microscopy (AC HAADF-STEM) and X-ray absorption spectroscopy (XAS) demonstrated that the single-atom Cu species were uniformly distributed and coordinated with two N atoms, and inductively coupled plasma mass spectrometry (ICP-MS) determined the Cu content of 1.45 wt% in Cu–N2/GN nanosheets. The coordinatively unsaturated Cu not only promoted the adsorption of CO2 on the catalyst surface, but also accelerated the electron transfer from Cu–N2 sites to *CO2. The Cu–N2/CN catalyst produced CO at a low overpotential, with a maximum FE of 81% at −0.50 V vs. RHE, and onset potential −0.33 V vs. RHE (Table 1). They found that the electronegative N atoms near the coordinatively unsaturated metal center further reduced the energy barrier, lowering the onset potential to −0.30 V vs. RHE [66]. Though we focus on Cu-based catalysts in this review, their effect on reducing the overpotential of CO2RR, especially for two-electron products, is inferior to catalysts with other metals. Zhang et al. prepared singly dispersed FeN5 sites supported on N-doped graphene with an additional axial ligand coordinated to FeN4 through thermal pyrolysis [52]. The AC HAADF-STEM images showed the atomically dispersed Fe and an absence of larger clusters in as-prepared catalysts. In electrochemical tests, the over-coordinated catalyst exhibited a high FE of 97% for CO production at low overpotential. DFT calculation disclosed that the additional coordination number weakened the *CO binding strength of FeN5, and facilitated the desorption of *CO, which changed the RDS and lowered the overpotential. The coordinated adjustment was also an effective way to modify the electronic structures of the metal center and enhance the catalytic performance. Yang et al. synthesized an S-doped Ni-SAC (Ni–N3S) by pyrolysis treatment [53]. The single-Ni-atom catalyst was prepared by pyrolysing a mixture of the amino acid (l-alanine or l-cysteine), melamine, and nickel acetate in argon, with the addition of a sulfur precursor (l-cysteine)The catalytic results showed that the S doping reduced the onset overpotential at only 70 mV, 100 mV lower compared to the Ni–N4 catalyst without S doping. Ni K-edge X-ray absorption near-edge structure (XANES) spectra indicated that the non-centrosymmetric ligand strength of Ni–N3S highly distorted the geometry, which was considered to promote the adsorption of reactants and intermediates, and reduce the overpotential. Cl and N dual-coordinated Mn-SAC ((Cl, N)-Mn/G) synthesized by Zhang et al. improved CO2RR, and the catalytic activity was obtained at low overpotential [67]. The d-band center of (Cl, N)-Mn/G was lower than that of MnN4, thus weakening the strong adsorption for *CO. DFT calculation proved that the energy barrier of the RDS (desorption of *CO) decreased from 1.64 eV for MnN4 to 0.65 eV for (Cl, N)-Mn/G. As there were four types of N atoms existing in SACs, different types of N atoms coordinated with the metal center and resulted in the variation of catalytic performance. Gu et al. prepared Fe3+-N-C coordinated with pyrrolic N, which displayed a CO partial current density of 94 mA cm−2 at an overpotential of 340 mV [54]. In this catalyst, the pyrrolic N coordination rendered the Fe3+/2+ reduction potential more negative than the Fermi level of the carbon support, leading to the stabilization of the Fe3+ ions in CO2RR conditions. Then, the stabilized Fe3+ induced faster CO2 adsorption and weaker CO absorption, which contributed to the high activity with low overpotential.
This entry is adapted from the peer-reviewed paper 10.3390/catal12080860