Multiple sclerosis (MS) is an inflammatory demyelinating autoimmune disease of the central nervous system (CNS) that typically manifests with episodes of new/recurrent neurological symptoms (i.e., relapses) followed by either complete remission or residual disability (relapsing–remitting, RR, course). Recent evidence of effectiveness of B cell-depleting monoclonal antibodies (mAbs) in MS prompted a partial revisitation of the pathogenetic paradigm of the disease, which has been, so far, considered a T cell-mediated autoimmune disorder. Although mechanisms underlying the efficacy of B cell-depleting mAbs in MS are not fully elucidated, they likely involve the impairment of pleiotropic B cell functions different from antibody secretion, such as their role as antigen-presenting cells. A potential impact of B cell-depleting mAbs on inflammation compartmentalized within the central nervous system was also suggested, but little is known about the mechanism underlying this latter phenomenon.
- multiple sclerosis
- B cell-depleting therapy
- monoclonal antibody
1. Introduction
2. Insights into MS Pathogenesis: Onset of Autoimmunity
2.1. Primary Autoimmune Response in the Peripheral Compartment
2.2. Secondary Autoimmune Response in the Central Nervous System
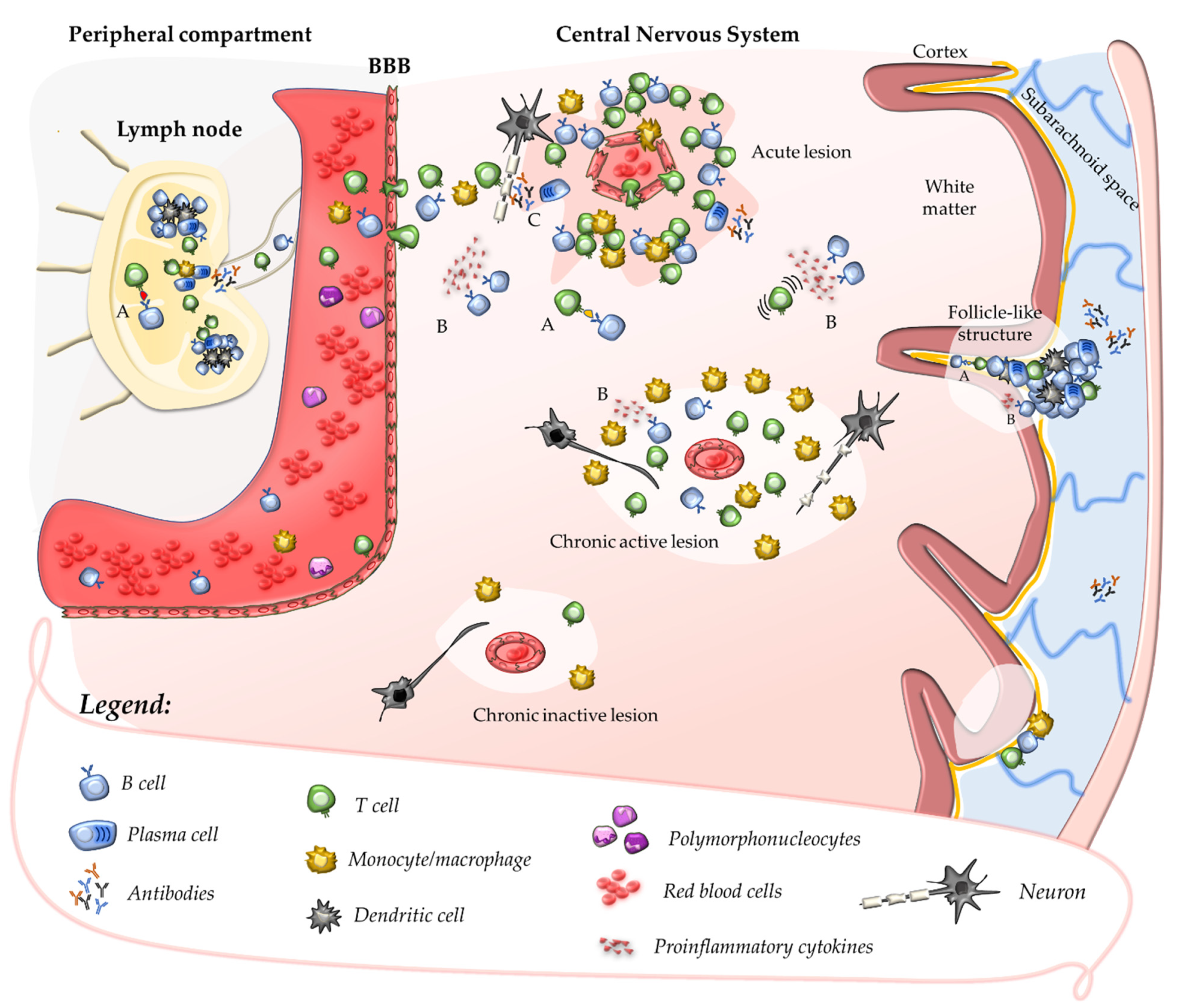
2.3. Contribution of B Cells and Humoral Response to Acute CNS Injury
This entry is adapted from the peer-reviewed paper 10.3390/jcm11154288
References
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636.
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286.
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2018, 9, 3116.
- Lassmann, H. Pathology and disease mechanisms in different stages of multiple sclerosis. J. Neurol. Sci. 2013, 333, 1–4.
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schroder, R.; Deckert, M.; Schmidt, S.; et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 2000, 192, 393–404.
- Junker, A.; Ivanidze, J.; Malotka, J.; Eiglmeier, I.; Lassmann, H.; Wekerle, H.; Meinl, E.; Hohlfeld, R.; Dornmair, K. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain 2007, 130, 2789–2799.
- Skulina, C.; Schmidt, S.; Dornmair, K.; Babbe, H.; Roers, A.; Rajewsky, K.; Wekerle, H.; Hohlfeld, R.; Goebels, N. Multiple sclerosis: Brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc. Natl. Acad. Sci. USA 2004, 101, 2428–2433.
- Markovic-Plese, S. Degenerate T-cell receptor recognition, autoreactive cells, and the autoimmune response in multiple sclerosis. Neuroscientist 2009, 15, 225–231.
- Montes, M.; Zhang, X.; Berthelot, L.; Laplaud, D.A.; Brouard, S.; Jin, J.; Rogan, S.; Armao, D.; Jewells, V.; Soulillou, J.P.; et al. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin. Immunol. 2009, 130, 133–144.
- Saxena, A.; Martin-Blondel, G.; Mars, L.T.; Liblau, R.S. Role of CD8 T cell subsets in the pathogenesis of multiple sclerosis. FEBS Lett. 2011, 585, 3758–3763.
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558.
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189.
- Amoriello, R.; Chernigovskaya, M.; Greiff, V.; Carnasciali, A.; Massacesi, L.; Barilaro, A.; Repice, A.M.; Biagioli, T.; Aldinucci, A.; Muraro, P.A.; et al. TCR repertoire diversity in Multiple Sclerosis: High-dimensional bioinformatics analysis of sequences from brain, cerebrospinal fluid and peripheral blood. EBioMedicine 2021, 68, 103429.
- Amoriello, R.; Greiff, V.; Aldinucci, A.; Bonechi, E.; Carnasciali, A.; Peruzzi, B.; Repice, A.M.; Mariottini, A.; Saccardi, R.; Mazzanti, B.; et al. The TCR Repertoire Reconstitution in Multiple Sclerosis: Comparing One-Shot and Continuous Immunosuppressive Therapies. Front Immunol. 2020, 11, 559.
- Gestri, D.; Baldacci, L.; Taiuti, R.; Galli, E.; Maggi, E.; Piccinni, M.P.; Vergelli, M.; Massacesi, L. Oligoclonal T cell repertoire in cerebrospinal fluid of patients with inflammatory diseases of the nervous system. J. Neurol. Neurosurg. Psychiatry 2001, 70, 767–772.
- Martin, R.; Jaraquemada, D.; Flerlage, M.; Richert, J.; Whitaker, J.; Long, E.O.; McFarlin, D.E.; McFarland, H.F. Fine specificity and HLA restriction of myelin basic protein-specific cytotoxic T cell lines from multiple sclerosis patients and healthy individuals. J. Immunol. 1990, 145, 540–548.
- Bielekova, B.; Sung, M.H.; Kadom, N.; Simon, R.; McFarland, H.; Martin, R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J. Immunol. 2004, 172, 3893–3904.
- Hellings, N.; Baree, M.; Verhoeven, C.; D’Hooghe, M.B.; Medaer, R.; Bernard, C.C.; Raus, J.; Stinissen, P. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J. Neurosci. Res. 2001, 63, 290–302.
- del Pilar Martin, M.; Cravens, P.D.; Winger, R.; Kieseier, B.C.; Cepok, S.; Eagar, T.N.; Zamvil, S.S.; Weber, M.S.; Frohman, E.M.; Kleinschmidt-DeMasters, B.K. Depletion of B lymphocytes from cerebral perivascular spaces by rituximab. Arch. Neurol. 2009, 66, 1016–1020.
- Sawcer, S.; Franklin, R.J.; Ban, M. Multiple sclerosis genetics. Lancet Neurol. 2014, 13, 700–709.
- Amato, M.P.; Derfuss, T.; Hemmer, B.; Liblau, R.; Montalban, X.; Soelberg Sorensen, P.; Miller, D.H.; Group, E.F.W. Environmental modifiable risk factors for multiple sclerosis: Report from the 2016 ECTRIMS focused workshop. Mult. Scler. 2017, 24, 590–603.
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768.
- Jersild, C.; Svejgaard, A.; Fog, T. HL-A antigens and multiple sclerosis. Lancet 1972, 1, 1240–1241.
- Ryan, S.O.; Cobb, B.A. Roles for major histocompatibility complex glycosylation in immune function. Semin. Immunopathol. 2012, 34, 425–441.
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836.
- International Multiple Sclerosis Genetics, Consortium; ANZgene; IIBDGC; WTCCC2. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365.
- Titus, H.E.; Chen, Y.; Podojil, J.R.; Robinson, A.P.; Balabanov, R.; Popko, B.; Miller, S.D. Pre-clinical and Clinical Implications of “Inside-Out” vs. “Outside-In” Paradigms in Multiple Sclerosis Etiopathogenesis. Front. Cell. Neurosci. 2020, 14, 599717.
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106.
- Harkiolaki, M.; Holmes, S.L.; Svendsen, P.; Gregersen, J.W.; Jensen, L.T.; McMahon, R.; Friese, M.A.; van Boxel, G.; Etzensperger, R.; Tzartos, J.S.; et al. T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity 2009, 30, 348–357.
- Haring, J.S.; Pewe, L.L.; Perlman, S. Bystander CD8 T cell-mediated demyelination after viral infection of the central nervous system. J. Immunol. 2002, 169, 1550–1555.
- Sospedra, M.; Martin, R. Immunology of Multiple Sclerosis. Semin. Neurol. 2016, 36, 115–127.
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301.
- ’t Hart, B.A.; Luchicchi, A.; Schenk, G.J.; Stys, P.K.; Geurts, J.J.G. Mechanistic underpinning of an inside–out concept for autoimmunity in multiple sclerosis. Ann. Clin. Transl. Neurol. 2021, 8, 1709–1719.
- Laman, J.D.; Weller, R.O. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J. Neuroimmune Pharmacol. 2013, 8, 840–856.
- Mundt, S.; Greter, M.; Flugel, A.; Becher, B. The CNS Immune Landscape from the Viewpoint of a T Cell. Trends Neurosci. 2019, 42, 667–679.
- Mapunda, J.A.; Tibar, H.; Regragui, W.; Engelhardt, B. How Does the Immune System Enter the Brain? Front. Immunol. 2022, 13, 805657.
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol. 2008, 67, 1113–1121.
- Kawakami, N.; Flügel, A. Knocking at the brain’s door: Intravital two-photon imaging of autoreactive T cell interactions with CNS structures. Semin. Immunopathol. 2010, 32, 275–287.
- Croxford, J.L.; Olson, J.K.; Miller, S.D. Epitope spreading and molecular mimicry as triggers of autoimmunity in the Theiler’s virus-induced demyelinating disease model of multiple sclerosis. Autoimmun. Rev. 2002, 1, 251–260.
- Scalfari, A.; Neuhaus, A.; Degenhardt, A.; Rice, G.P.; Muraro, P.A.; Daumer, M.; Ebers, G.C. The natural history of multiple sclerosis: A geographically based study 10: Relapses and long-term disability. Brain 2010, 133, 1914–1929.
- Filippi, M.; Rocca, M.A.; Ciccarelli, O.; De Stefano, N.; Evangelou, N.; Kappos, L.; Rovira, A.; Sastre-Garriga, J.; Tintorè, M.; Frederiksen, J.L. MRI criteria for the diagnosis of multiple sclerosis: MAGNIMS consensus guidelines. Lancet Neurol. 2016, 15, 292–303.
- Absinta, M.; Sati, P.; Schindler, M.; Leibovitch, E.C.; Ohayon, J.; Wu, T.; Meani, A.; Filippi, M.; Jacobson, S.; Cortese, I.C.; et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J. Clin. Investig. 2016, 126, 2597–2609.
- Weber, M.S.; Hemmer, B.; Cepok, S. The role of antibodies in multiple sclerosis. Biochim. Biophy. Acta 2011, 1812, 239–245.
- Li, R.; Patterson, K.R.; Bar-Or, A. Reassessing B cell contributions in multiple sclerosis. Nat. Immunol. 2018, 19, 696–707.
- Kabat, E.A.; Moore, D.H.; Landow, H. An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins. J. Clin. Investig. 1942, 21, 571–577.
- Correale, J.; de los Milagros Bassani Molinas, M. Oligoclonal bands and antibody responses in multiple sclerosis. J. Neurol. 2002, 249, 375–389.
- Gaitan, M.I.; Maggi, P.; Wohler, J.; Leibovitch, E.; Sati, P.; Calandri, I.L.; Merkle, H.; Massacesi, L.; Silva, A.C.; Jacobson, S.; et al. Perivenular brain lesions in a primate multiple sclerosis model at 7-tesla magnetic resonance imaging. Mult. Scler. 2014, 20, 64–71.
- Hart, B.A.; Massacesi, L. Clinical, pathological, and immunologic aspects of the multiple sclerosis model in common marmosets (Callithrix jacchus). J. Neuropathol. Exp. Neurol. 2009, 68, 341–355.
- Genain, C.P.; Cannella, B.; Hauser, S.L.; Raine, C.S. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med. 1999, 5, 170–175.
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717.
- Cencioni, M.T.; Mattoscio, M.; Magliozzi, R.; Bar-Or, A.; Muraro, P.A. B cells in multiple sclerosis-from targeted depletion to immune reconstitution therapies. Nat. Rev. Neurol. 2021, 17, 399–414.
- Lovato, L.; Willis, S.N.; Rodig, S.J.; Caron, T.; Almendinger, S.E.; Howell, O.W.; Reynolds, R.; O’Connor, K.C.; Hafler, D.A. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain 2011, 134, 534–541.
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174.
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134, 2755–2771.
- Wang, J.; Jelcic, I.; Muhlenbruch, L.; Haunerdinger, V.; Toussaint, N.C.; Zhao, Y.; Cruciani, C.; Faigle, W.; Naghavian, R.; Foege, M.; et al. HLA-DR15 Molecules Jointly Shape an Autoreactive T Cell Repertoire in Multiple Sclerosis. Cell 2020, 183, 1264–1281.e20.