2. Physiology of UA
2.1. Source of UA
Elevated serum UA levels are exacerbated by diets high in fat, protein, or nucleic acid components. Among these products, purine nucleic acid ingestion exhibited the greatest dietary influence on blood UA levels [
10,
11]. The majority of the endogenous UA is produced through purine metabolism. A healthy male on a purine-free diet usually has a total purine generation of approximately 1200 mg, and a female of approximately 600 mg, which includes endogenous (daily synthesis rates of approximately 300–400 mg) and exogenous purines (usually approximately 300 mg per day influenced by dietary content) [
10,
12,
13]. Although the production and catabolism of purines are relatively constant between 300 mg and 400 mg per day, the relationship between diet and serum UA are more complex than simple purine intake. As mentioned previously, diet content other than purine also influences UA generation. For instance, provisions with a high fructose content, beer, and soft drinks with high-fructose corn syrup influence fructokinase activation, which leads to rapid UA production [
14,
15,
16].
2.2. UA Clearance
In most mammalian species, UA is not the end product of the purine nucleic acid metabolic pathway. UA is further metabolized to the more soluble allantoin by uricase and readily excreted in the urine. However, humans lack the functional uricase enzymes, making UA the final degradation product of the metabolic pathway [
18]. In humans, approximately two-thirds of urate is excreted in the urine with normal uricosuria levels of approximately 620 mg/day in adults, with the remainder largely excreted via the gastrointestinal tract [
19]. In the kidneys, nearly all of the serum urate is filtered by the glomeruli, but approximately 91–95% is reabsorbed in the proximal tubule via renal tubule transporters, such as urate anion transporter 1 (URAT1, encoded by the SLC22A12 gene) [
20,
21]. Eventually, approximately 3–10% of the filtered urate appears in the urine after the glomerular filtration, proximal tubule reabsorption, secretion, and post-secretory reabsorption. Thus, hyperuricemia may also lead to hyperuricosuria, defined as a 24 h urinary excretion of over 800 mg in men and 750 mg in women. UA within the urine tends to crystallize when the urine pH is low; therefore, hyperuricosuria combined with acidic urine and low urine volume might result in urate stone formation. Previous reports have pointed out that DM patients had significantly higher urine UA levels and a lower urine pH compared to the nondiabetic population, making DM patients prone to develop urate stones [
22,
23]. Not only the renal transport system influenced the urate metabolites, but the decreasing of gastrointestinal urate excretion also increased the risk of hyperuricemia. For example, the ABCG2, a high-capacity urate exporter, dysfunction had been reported to increase the risk of hyperuricemia due to a decrease in intestinal urate excretion [
24].
2.3. Genetic Variations Influence the Serum UA Level
Hyperuricemia should be considered as a combined result of UA overproduction and underexcretion by the kidneys; therefore, all of the genetic variations involved in UA metabolism and elimination could contribute to hyperuricemia.
Several single-locus mutations are known to cause hyperuricemia via UA overproduction. Hypoxanthine-guanine phosphoribosyltransferase (HPRT) is an enzyme that recycles hypoxanthine and guanine. HPRT malfunction results in excessive UA production. Lesch–Nyhan syndrome, an X-linked disorder of HPRT deficiency, is characterized by UA overproduction resulting in precocious hyperuricemia, UA nephrolithiasis, spasticity, and mental retardation [
25]. Phosphoribosylpyrophosphate (PRPP) synthetase takes part in the very first step in the de novo synthesis of purines and increasing the activity of PRPP synthetase leads to a purine overproduction and subsequent serum UA elevation. PRPP synthetase overactivity and mutation of the PRPP synthetase 1 gene on Xq22-24 result in hyperuricemia, hyperuricosuria, dysmorphic deafness, and severe mental retardation [
26,
27]. Treatment with allopurinol, which reduces UA production by inhibiting XO, shows promising effects in controlling hyperuricemia, UA nephrolithiasis, and the metabolic complications of these two disorders.
3. Hyperuricemia and Renal Disease
According to cross-sectional case-control studies, hyperuricemia and gout have been reported to be comorbid with multiple diseases, including coronary artery disease, hypertension, type 2 DM, or CKD, and are independent risk factors for premature death [
35,
36,
37]. Gout is associated with all the components of the metabolic syndrome, and the prevalence of these comorbidities increases with the gout duration [
38]. However, it is still unclear whether elevated serum UA levels are a causal factor in the development of these comorbidities, or whether hyperuricemia and gout share similar risk factors with these disorders.
As mentioned previously, the relationship between hyperuricemia and renal impairment was investigated as early as the 1960s via a postmortem analysis. At the time, the presence of kidney disease in gout patients was considered to be one of the complications of gout and was attributed to the presence of urate crystals in the kidney, leading to the extensive use of the term gouty nephropathy. However, in the 1980s, the viewpoint of the relationship between gout and renal impairment led us to propose that both of these diseases were the consequence of hypertension, causing an initial decline in the renal function and the subsequent elevation of serum UA levels due to decreased urate excretion via the kidneys. Based on this theory, the treatment of hyperuricemia in patients with kidney disease was only recommended to manage gout, renal stones, or, in rare cases, the tumor lysis syndrome. The treatment of asymptomatic hyperuricemia was not recommended at that time.
4. Mechanism
According to experimental studies focused on the identification of mechanistic pathways, several mechanisms exist that might explain why serum urate or urinary UA might cause CKD. By understanding these important pathophysiological processes, investigators can design clinical trials more specifically and interpret the outcomes of clinical trials more accurately.
4.1. Crystalline Effects
The crystalline effect was discovered initially and is the most widely known negative impact of UA. Glomerular ultrafiltration can be enriched with minerals, proteins, or drug metabolites, which tend to supersaturate under volume depletion or specific urine pH levels. Tumor lysis syndrome, heat stress, and rhabdomyolysis can induce an abundant UA release from cells and transiently increase serum UA levels. The acute elevation of serum UA levels subsequently increases urinary urate levels and precipitates urate crystal formation in the renal tubular lumen, especially in acidic urine or dehydration status [
50,
51]. In addition to these disorders, certain types of purine ingestion also lead to transient increases in serum UA levels with subsequent uricosuria and crystal formation [
7,
10]. The formation of crystals induced by acute supersaturation states leads to crystal-associated tubular cell injury and renal function impairment.
In contrast to acute events, long-standing moderate supersaturation can promote crystal formation and tubule obstruction over extended periods of time and even lead to tissue remodeling [
52,
53,
54]. Sellmayr and his colleagues had probed the role of crystal-induced inflammation and macrophages in the pathology of progressive CKD via a novel mouse model, which revealed that UA crystal granulomas develop late in chronic UA crystal nephropathy and contribute to CKD progression via trigger M1-like macrophage-related interstitial inflammation and fibrosis [
55]. The long-term elevation of urine UA levels constitutes an endogenous danger signal that activates the inflammasome-mediated inflammatory response [
56,
57,
58]. A previous study revealed that soluble UA enhances pyrin domain containing 3 (NALP3) expression, caspase-1 activation, and interleukin-1β and intercellular adhesion molecule-1 levels in the renal proximal tubule cells in a Toll-like receptor 4-dependent manner (
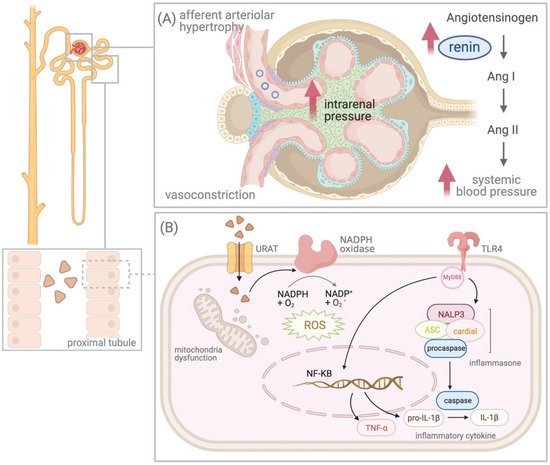
Figure 1) [
57]. Another study showed that UA induces renal inflammation via the activation of the nuclear factor-κB signaling pathway [
58,
59,
60]. Ryu et al. also reported that the uptake of UA by tubular epithelial cells might induce epithelial-to-mesenchymal transition by decreasing E-cadherin synthesis and increasing E-cadherin degradation [
11]. By inducing the inflammatory activation and cell remodeling, elevated urinary UA levels result in chronic renal function decline.
Figure 1. The effects of hyperuricemia on the kidney. (A) Stimulation of the renin–angiotensin–aldosterone system and activation of the vasoconstrictors due to hyperuricemia results in elevated intrarenal pressure and afferent arteriolar hypertrophy. (B) Soluble uric acid (UA) activates inflammasome pyrin domain containing 3 (NALP3)-induced inflammasome-mediated inflammatory response and intracellular UA increases oxidative stress via the stimulation of nicotinamide adenine dinucleotide (NADPH), leading to mitochondrial dysfunction. Abbreviations: Ang, Angiotensin; URAT, urate anion transporter; ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; IL, interleukin; NADPH, nicotinamide adenine dinucleotide; NF-κB, nuclear factor-kappa B; TLR, Toll-like receptor; TNF, tumor necrosis factor; ROS, reactive oxygen species.
4.2. UA and Renin–Angiotensin–Aldosterone System (RAAS)
Mazzali et al. revealed that rats with hyperuricemia developed hypertension after 3 weeks, which can be prevented or improved through the administration of allopurinol or benziodarone. According to the findings of this animal study, it was concluded that hyperuricemia itself can cause hypertension and renal injury via a crystal-independent mechanism, which can lead to hypertension and renal function impairment through the stimulation of the RAAS and inhibition of nitric oxide synthesis [
7,
46,
61,
62,
63]. The following experiments also supported the theory that UA first activates the prorenin receptors in proximal tubule cells and then stimulates the intrarenal RAAS, increasing renal renin expression and elevating the serum aldosterone and intracellular angiotensin II levels [
62,
64,
65]. This effect is not only revealed in animal models. One randomized, double-blind, placebo-controlled, crossover trial reported that allopurinol can markedly reduce plasma renin activity and decrease the blood pressure level in adults newly diagnosed with hypertension [
66]. Talaat and his colleagues revealed the minimal effects on kidney function and blood pressure level in CKD patients who stopped allopurinol therapy but maintained an angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker. However, CKD patients who did not receive RAAS blockers showed marked deterioration in renal function and blood pressure after the cessation of allopurinol therapy. Thus, it was concluded that asymptomatic hyperuricemia has a deleterious effect on the progression of CKD and hypertension, which can be blocked by RAAS blockers [
67].
In addition to the RAAS, UA activates other vasoconstrictors, such as endothelin and thromboxane, and suppresses vasodilatory pathways such as NO [
41,
68]. The result of the use activation vasoconstrictors and suppression of the vasodilators causes systemic and renal vasoconstriction, thereby reducing the renal plasma flow. The persistently elevated intrarenal pressure causes afferent arteriolar hypertrophy, which impairs the renal autoregulation system and weakens the afferent vasoconstrictive response to systemic pressure (
Figure 1). The transmission of systemic blood pressure to the glomeruli promotes CKD progression [
69]. Rodriguez-Iturbe et al. revealed that persistent hypertension induced the development of low-grade kidney interstitial inflammation involving T cells and macrophages, which continued even after the factors inducing hypertension were initially removed. The innate and adaptive immunity activated by persistent hypertension initially further results in inflammation-induced impairment of the pressure–natriuresis relationship, dysfunction of vascular relaxation, and sympathetic nervous system overactivity [
70]. Afferent arteriolar hypertrophy, interstitial inflammation, and impairment of the pressure–natriuresis relationship might explain the phenomenon that hypertension induced by hyperuricemia is initially responsive to ULT but fails to respond to ULT once renal vascular disease, interstitial inflammation, and salt-sensitive hypertension have developed [
71].
4.3. Intracellular Effect of UA
Although most previous gout studies have mentioned the extracellular effects of UA, mainly the inflammatory response or toxic effects via crystalline deposition in joint spaces or renal tubules, more recent experimental studies have shown that the primary metabolic and renal effects of UA are mediated by intracellular urate [
72,
73]. Intracellular UA increases intracellular oxidative stress via the stimulation of nicotinamide adenine dinucleotide (NADPH) oxidase, leading to mitochondrial dysfunction. Intracellular oxidative stress then promotes inflammatory cytokine activation and triggers vascular smooth muscle cell proliferation, which further induces a vasoactive response [
41,
74]. It seems reasonable that the elevation of the extracellular effect of the UA level will increase intracellular UA, but endogenous production of UA, for example, fructose metabolism via XO, could elevate the intracellular UA levels, even when the serum urate levels are within the normal range [
75]. This mechanism might explain the synergistic effects of allopurinol and captopril on the blood pressure level in animal models, which cannot be well explained by the RAAS inhibition mechanism alone [
76]. Based on these studies, clinicians realized that ULT might provide additional metabolic protection intracellularly beyond the blockade of the RAAS. To date, many studies have used allopurinol or ULT as an add-on therapy to RAAS blockers, and many studies aim to reveal the possible effect of inhibiting cellular UA uptake by uricosuric agents or using antioxidants to eliminate the intracellular oxidative stress induced by urate.
5. Treatment and Evidence of Clinical Trials
Several ULT drugs have been approved in the past few years, which can be classified into three main categories: drugs inhibiting UA synthesis (xanthine oxidase inhibitors (XOi), for example, allopurinol, febuxostat, and topiroxostat); drugs increasing UA excretion (uricosurics, for example, benzbromarone and lesinurad); and drugs enabling systemic metabolic hydrolysis of UA (urolytics, for example, rasburicase and pegloticase). Considering that the main mechanism of uricosuric agents is to increase renal urate excretion, these agents might increase the risk of adverse renal events due to the elevated urinary UA levels by increasing the renal clearance of UA [
21,
77]. Moreover, urolytic agents are only approved for chronic refractory gout with tophaceous deformities or hyperuricemia induced by tumor lysis syndrome and are prescribed through intravenous administration. Clinical studies investigating the role of ULT in CKD prevention have mostly focused on XOis. XOis inhibit XO to reduce the endogenous production of UA and subsequently lower the serum UA levels and can be categorized into two main classes: classical purine analogs (allopurinol) and nonpurine analog compounds (febuxostat and topiroxostat) [
63].