Lysine-specific histone demethylase 3 (KDM3) subfamily proteins are H3K9me2/me1 histone demethylases that promote gene expression. The KDM3 subfamily primarily consists of four proteins (KDM3A−D). All four proteins contain the catalytic Jumonji C domain (JmjC) at their C-termini, but whether KDM3C has demethylase activity is under debate. In addition, KDM3 proteins contain a zinc-finger domain for DNA binding and an LXXLL motif for interacting with nuclear receptors. Of the KDM3 proteins, KDM3A is especially deregulated or overexpressed in multiple cancers, making it a potential cancer therapeutic target. However, no KDM3A-selective inhibitors have been identified to date because of the lack of structural information. Uncovering the distinct physiological and pathological functions of KDM3A and their structure will give insight into the development of novel selective inhibitors.
- KDM3A
- histone H3K9 demethylase
- KDM3 inhibitor
- anti-cancer agent
- epigenetic agent
1. Introduction
Histone modifications are dynamically controlled by chromatin-modifying enzymes that read specific positions and add or remove corresponding covalent modifications. Among the several different types of enzymes, lysine-specific histone methyltransferases catalyze reversible mono-, di-, and tri-methylation via an S-adenosyl-L-methionine (SAM)-dependent mechanism [1] [2]. Conversely, lysine-specific histone demethylases (KDMs) remove these methylation marks [3], contributing to the plasticity of covalent histone modifications. Multiple functional domains are essential for KDM activity. Most KDMs contain a catalytic Jumonji C (JmjC) domain that also functions as a binding pocket for catalytically necessary cofactors [4]. As a result, KDMs are divided into two classes according to the type of cofactor used in their catalytic mechanisms, namely flavin adenine dinucleotide (FAD) [5] or α-ketoglutarate (α-KG also called 2-oxoglutarate (2-OG)) with iron Fe(II) [6]. FAD-dependent enzymes form the KDM1 or Lys-specific demethylase (LSD) subfamily [5], whereas α-KG and Fe(II)-dependent enzymes form JmjC containing demethylase [6][7]. Compared with the restricted catalytic activity of KDM1 at mono- and dimethyl Lys residues, the second class of demethylases is capable of removing all three possible methylation states of methylated Lys residues [8]. Of the 30 known KDMs, 28 of the enzymes belong to the JmjC-KDM class [9], which is divided into five subfamilies: KDM2/7, KDM3, KDM4, KDM5, and KDM6.
Human KDM3 is also known as JMJD1 (Jumonji domain-containing 1) and JHDM2 (Jumonji C domain-containing histone demethylase 2). The KDM3 subfamily encompasses four members (Figure 1): KDM3A, KDM3B, KDM3C, and KDM3D. Of these proteins, the enzymatic activity of KDM3C remains controversial [9,10]. KDM3A and KMD3B catalyze the demethylation of transcriptionally repressive mono- and di-methylated histone H3 lysine 9 (H3K9me1/me2) in vitro and in vivo with a preference for dimethylated residues, thereby mediating transcriptional activation [10][11]. KDM3D/hairless (HR) protein demethylates H3K9me1 in vitro and in vivo, and it can also weakly demethylate H3K9me2 in vivo [12]. The KDM3 proteins share a common domain architecture consisting of a JmjC domain at the C-terminus, a catalytic histone demethylase domain [13] and non-catalytic C2HC2 zinc (Zn) finger, and LXXLL (where L is leucine and X is any amino acid) motifs for interacting with nuclear hormone receptors (NRs). The Zn-finger domain of KDM3A is involved in determining substrate specificity [10][11] but its exact mechanism is unknown. Additionally, the JmjC and the Zn-finger domains are required for forming a homodimer of KDM3A, which is important for its functions [11][14]. Further studies are needed to determine whether other KDM3 proteins undergo homodimerization.
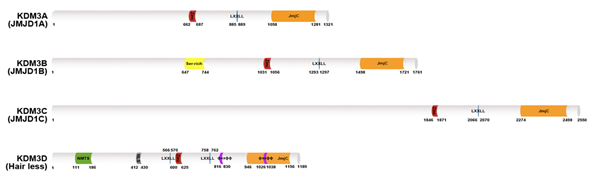
Figure 1. Lysine-specific histone demethylase 3 (KDM3) demethylase protein structures. The KDM3A–D protein architecture consists of a common C2HC4-type zinc-finger-like motif (red color) LXXLL motif (blue color) and one Jumonji C domain (JmjC) domain (orange color) in the C-terminal catalytic site. KDM3B contains a serine-rich region (yellow color). KDM3D also possesses a nuclear matrix targeting signal (NMTS; green color) a nuclear localization signal (NLS; gray color) and two ϕXXϕϕ motifs (purple color) that serve as thyroid hormone receptor interaction domains.
KDM3 proteins are deregulated in skin, hair, and cardiovascular diseases and multiple cancers, including breast, prostate, and colon cancers, and lymphomas, and thus, they have emerged as potential broad-spectrum therapeutic targets. Each of these KDM3 proteins plays differential yet redundant functions in both physiological and pathological processes (Figure 2). Among them, KDM3A was first cloned and characterized as a mouse male germ cell-specific transcript in 1991 [15]. KDM3A demethylates H3K9me1/me2 [11] to increase gene transcription and plays key roles in spermatogenesis [16], energy metabolism [17], stem cell regulation [18], and sex determination [19]. Moreover, increasing evidence has indicated the key roles of KDM3A in promoting cancer progression. (1) KDM3A is frequently upregulated in multiple cancers and elevated KDM3A levels are associated with worse cancer prognosis [20][21]. (2) Depletion of KDM3A significantly inhibits colony formation [22][23], cell proliferation [24][25] and xenograft tumor formation [26][27]. (3) The depletion of KDM3A substantially suppresses migration, invasion, and metastasis [28][29]. (4) KDM3A regulates oncogene expression by binding NRs (e.g., AR [20][30], ER [31][32][33], HIF-1α [24][25]) and modulating downstream signaling (e.g., Wnt/β-catenin signaling [26][34] and Hippo signaling [27]).
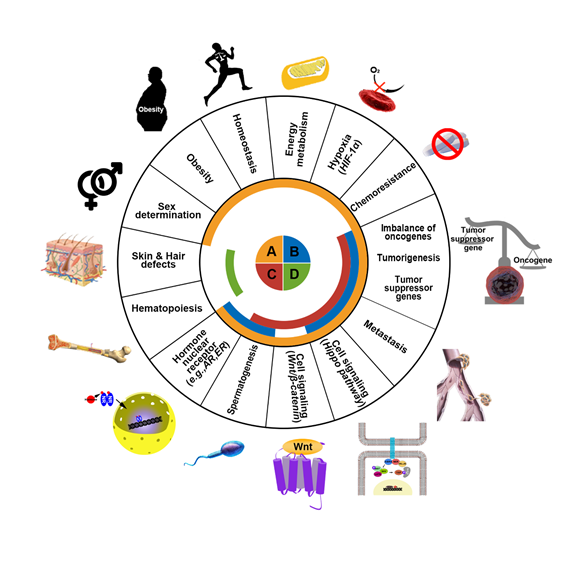
Figure 2. Schematic representation of the major functions of KDM3 in humans. The KDM3 family consists of four proteins: KDM3A (orange) KDM3B (blue) KDM3C (red) and KDM3D (green). These proteins regulate gene expression and chromatin dynamics by demethylating H3K9me1/2. KDM3 family proteins normally function in energy metabolism, homeostasis, obesity, sex determination, hematopoiesis, hormone nuclear receptor signaling, spermatogenesis, and cell signaling pathways. However, dysregulation in KDM3 expression or function promotes skin and hair defects and tumorigenic processes such as cell survival under hypoxia, malfunction of tumor suppressor genes, induction of oncogenes, development of chemoresistance, and metastasis in cancer patients.
2. Altered Expression and Functions of KDM3A in Cancer
Numerous studies demonstrated that abnormal KDM3 expression promotes the progression of cancer, as substantiated by the number of published studies shown in Table 1. KDM3A and KDM3B are well known to be tightly related to cancer, whereas KDM3C and KDM4D are relatively unstudied in cancer development. Among the four KDMs, this review focuses on the relatively well-studied association between KDM3A and various cancers.
2.1. Prostate Cancer (PCa)
KDM3A is overexpressed in PCa [20][30][35]. In prostate adenocarcinoma, androgen-dependent signaling plays a key role in tumorigenesis [20][30]. Ligand bound androgen receptor (AR) translocates into the nucleus upon dimerization, recruiting coactivators to androgen response elements (AREs) to regulate gene expression. As an AR coactivator, KDM3A is significantly enriched in AREs, and it alters the expression of androgen response genes through demethylation [20][30]. Using gene set enrichment analysis, it was identified that overexpressed KDM3A posttranscriptionally activates androgen signaling and cancer metabolic pathways, such as hypoxia and glycolytic pathways, thus upregulating oncogene expression. Moreover, in an orthotopic prostate tumor model using CWR22Rv1 cells with permanently activated AR, KDM3A knockdown disrupts tumor formation [30], highlighting the importance of KDM3A in androgen signaling and carcinogenesis.
In addition to its role in the androgen signaling pathway, KDM3A also regulates alternative splicing of AR pre-mRNA, generating AR variant-7 (AR-V7) [36]. AR-V7 is one of the major mechanisms through which patients with PCa develop resistance to androgen deprivation therapy, which blocks AR activity. However, in patients with castration-resistant PCa (CRPC), AR activity is restored through alternative splicing. KDM3A facilitates the recruitment of spliceosome components and binds to heterogeneous nuclear ribonucleoprotein F, a splicing regulator, to create constitutively active AR-V7. Knockdown of KDM3A in Rv1 and LN95 cells resulted in reduced protein and AR-V7 mRNA levels. In fact, tissue microarray demonstrates that KDM3A promotes AR-V7 expression in certain human PCa tissues. In the KDM3A-knockdown Rv1 xenograft prostate tumor model, tumor weight is decreased by 13-fold, whereas no tumor formation occurs in castrated mice.
KDM3A interacts with and regulates the transcription of both AR and hypoxia-inducible factor 1α (HIF-1α) [24][25]. KDM3A serves as a transcriptional coactivator of HIF1α and AR in the context of hypoxia. Hypoxia increases the occupancy of HIF-1α and KMD3A on the gene encoding prostate-specific antigen (PSA), a secreted chymotrypsin-like serine protease A that cleaves proteins that function in cell proliferation and metastasis. Chromatin immunoprecipitation (ChIP) assay results confirmed that HIF-1α, together with AR, recruits endogenous KDM3A to PSA enhancers. Furthermore, the occurrence of H3K9me1 and H3K9me2, both of which are substrates of KDM3A, on PSA enhancers is reduced under hypoxia, further supporting the idea that KMD3A promotes PCa progression through the epigenetic regulation of PSA by interacting with HIF-1α.
In addition to AR and HIF-1α, c-Myc is posttranscriptionally controlled by KDM3A, leading to cancer proliferation and survival [20][21]. Overexpression of c-Myc is associated with poor prognosis and recurrence of PCa. Knockdown of KDM3A significantly inhibits c-Myc and AR activities. Conversely, double depletion of KDM3A and AR fails to reduce c-Myc levels, whereas KDM3A knockdown without changes of AR levels successfully decreases c-Myc mRNA transcription [21]. KDM3A also regulates HUWE1, an E3 ubiquitin ligase known to target Myc increases the stability of c-Myc [20]. KDM3A inhibits HUWE1/c-Myc interaction and prevents the HUWE1-dependent degradation of c-Myc, thus increasing c-Myc levels and PCa cell proliferation. Furthermore, the expression levels of c-Myc are positively correlated with those of KDM3A in certain PCa subset specimens. KDM3A depletion strongly inhibits PCa cell growth, so KDM3A can serve as a potential target in the development of novel drugs for treating PCa.
2.2. Breast Cancer (BCa)
KDM3A is overexpressed in BCa tissues. The Cancer Genome Atlas (TCGA) human BCa database (n = 729) shows that patients with BCa and high KDM3A expression have a three-fold higher risk of death than those with normal or low KDM3A expression [37]. Two-thirds of patients with BCa express ER and require ER-mediated transcriptional activation for cancer survival [38]. Similar to KDM4B, KDM3A is a well-known key ER coregulatory protein in ER signaling [31][32][33]. KDM3A acts as a positive regulator of ER, and its absence impairs ER recruitment to the cis-regulatory elements of the target gene promoters. Global gene expression analysis demonstrated that KDM3A regulates approximately 42% of ER target genes [32]. KDM3A depletion using siRNA notably downregulates ER target genes GREB1 and pS2 regardless of the presence of the ER ligand, β-estradiol (E2). The absence of KDM3A elevates H3K9me2 levels at both GREB1 and pS2 estrogen response elements (EREs) even in the presence of E2, failing to remove the repressive marks of these loci. Additionally, KDM3A knockdown abrogates the recruitment of ER to enhancer regions of ER target genes such as CCND1 (Cyclin D1), MYC, and XBP1, thereby highlighting the importance of KDM3A in ER signaling [32][39]. Furthermore, BCa cells expressing catalytically inactive KDM3A exhibit downregulation of cell cycle regulatory genes and increased proliferation, confirming the role of KDM3A in BCa progression [32].
In addition to its single role as a coactivator, KDM3A also cooperates with KDM4B via an auto-regulatory loop to regulate the functions of ER by facilitating the recruitment of other ER coactivating proteins and ER transcriptional complexes to the EREs [33]. Both KDM3A and KDM4B are enriched at EREs of ER target genes, such as pS2, GREB1, and CCND1. Notably, the chromatin occupancy of KDM3A is depleted by the absence of KDM4B and vice versa, strongly suggesting that these demethylases need each other to facilitate their association with chromatin. However, the mRNA and protein levels of KDM3A and KDM4B exhibit different patterns. Although depletion of KDM4B downregulates KDM3A mRNA and protein levels in certain BCa cells, KDM4B protein expression is not affected by KDM3B knockdown in BCa cells, suggesting a regulatory mechanism in which KDM4B regulates KDM3A expression.
KDM3A is responsible for the chemoresistance and stemness in BCa by removing Lys372me1 on p53 and H3K9me2 [28] and controlling homeobox protein Hox-A1 (HOXA1) expression [31]. Methylation of p53 at Lys372 by SET9 histone lysine N-methyltransferase activates p53, causing it to enter and localize in the nucleus, in which it activates its target genes [28]. KDM3A inhibits such activity of p53 by demethylating Lys372, thus hampering p53-mediated pro-apoptosis and resistance to paclitaxel and cisplatin. Also, KDM3A directly associates with activated CDC42 kinase 1 (ACK1), a non-receptor tyrosine kinase that functions in tumorigenesis, resistance, and metastasis and acts as an integrator of various signaling pathways such as HER2 signaling. Further explanation of such pathway is stated in the review.
Many studies demonstrated that KDM3A is associated with the progression of BCa metastasis [28][29][32] by removing methylation marks on pro-invasive genes. KDM3A promotes the metastasis of BCa by interacting with a Brahma related gene 1 (BRG1), a mammalian chromatin remodeling complex, on the Mucin 1 (MUC1) promoter to remove repressive methylation marks and activate pro-invasive gene transcription [29]. BRG1 contributes to BCa metastasis via tumor necrosis factor-α (TNF-α) and interferon-gamma (IFN-γ) induced transcription of MUC1, which is a pro-metastatic oncoprotein that is aberrantly elevated in highly malignant BCa due to transcriptional upregulation. Such upregulation of MUC1 is attributed to the interaction of KDM3A with and recruitment by BRG1 on the MUC1 promoter in a signal transducer and activator of transcription 1 (STAT1)- and p65-dependent manner. Targeting KDM3A in patients with BCa can be a powerful strategy to overcome drug resistance and therefore cancer recurrence.
2.3. Colon Cancer
Similar to the abnormally elevated expression of KDM3 in PCa, KDM3A is also overexpressed in colon cancer, and its levels are increased in colorectal cancer (CRC) metastatic lesions [26][34][40][41]. According to TCGA data, KDM3A is significantly overexpressed in colon adenocarcinomas, and its expression is correlated with reduced survival rates and high rates of recurrence. KDM3A is present in both the cytoplasm and nucleus in CRC and head and neck cancers, but it is rarely detected in the nuclear matrix and heterochromatin regions [40][42]. Downregulation of KDM3A in HCT116 CRC cells inhibits the clonogenic activity of CRC cells [40][43], arrests cell cycle progression, suppresses CRC cell proliferation and migration, and reduces xenograft tumor formation [26][27]. Moreover, as identified through transcriptome analysis, KDM3A downregulation stimulates p53- and transforming growth factor-β1 (TGF-β1)-related tumor suppressor pathways and inhibits c-Myc- and N-myc-driven oncogenic pathways.
Hyper-activated Wnt/β-catenin signaling is associated with CRC recurrence, development of drug resistance [26][34] and metastasis [27][41]. Among KDM3 family proteins, KDM3A and KDM3B play crucial roles in activating the Wnt signaling pathway [34]. The absence of KDM3A or KDM3B inhibits the Wnt target genes AXIN2 and DKK1. Moreover, both KDM3A and KDM3B directly interact and recruit β-catenin [26] in HCT116 CRC cells and specifically remove H3K9me2 from the Wnt target gene promoters, controlling the tumorigenic ability of human colorectal CSCs [26][34]. Furthermore, the expression levels of KDM3A positively correlate with β-catenin target genes in CRC specimens from TCGA data and predictive of worse cancer outcomes.
Recently, it was found that the oncogenic role of KDM3A is regulated by acetylation on enhancers of Hippo target genes [44] and PHF5A [27]. KDM3A recruits p300 to Hippo target gene enhancers and facilitates the interaction between TEA domain transcription factor 1 (TEAD1), a Hippo pathway transcription factor, and yes-associated protein 1 (YAP1) by demethylating H3K9me1/2 on YAP1 enhancers [44]. As activated in the Hippo pathway, upon starvation, PHF5A, a component of U2 small nuclear ribonucleoproteins (snRNPs) of the spliceosome, is acetylated at Lys29 by p300 [27]. This acetylation enhances the interaction of PHF5A with U2 snRNPs and stabilizes KDM3A mRNA by reducing the abnormal retention of KDM3A intron 3, which carries a stop codon. Notably, PHF5A Lys29 acetylation and KDM3A upregulation are correlated with the poor prognosis of CRC. Thus, PHF5A acetylation induces the stabilization of KMD3A mRNA and enhances its expression, in which the protein consequently activates signaling pathways that promote CRC.
In addition to its role in activating Wnt/β-catenin and Hippo signaling pathways in colon cancers, KDM3A activates the gene transcription of 15-lipoxygenase-1 (15-LOX-1), which is silenced in cancer cells [45]. 15-LOX-1 regulates terminal cell differentiation and inflammation by producing multiple inflammation-regulatory lipid signaling mediators. In CRC cells, 15-LOX-1 is abnormally downregulated, but re-expression of 15-LOX-1 inhibits cell growth and restores its function in cell differentiation and apoptosis. Overall, these findings support the indispensable function of KDM3A in colonic tumorigenesis, thus illuminating its potential as a novel therapeutic target for inhibiting cancer growth.
2.4. Lung Cancer
KDM3A is suggested to aid lung adenocarcinoma cells in evading the immune system of patients by upregulating forkhead box P3 (Foxp3) expression in regulatory T cells (Tregs) [46]. Foxp3 expression, controlled by activated Toll‑like receptor 4 (TLR4), enhances immunosuppressive functions of Tregs by inducing inhibitory cytokine secretion and facilitate lung cancer cells to escape the immune system. Upregulation of TLR4 significantly increases the expression of KDM3A in A549 lung adenocarcinoma cells, which subsequently controls Foxp3 transcription. Knockdown of KDM3A in normally active TLR4 lung adenocarcinoma cells reduces Foxp3 expression and downstream cytokines such as TGF-β1 and interleukin-35 (IL-35), as well as heme oxygenase 1. Furthermore, the colocalization of KDM3A and Foxp3 and suppressed luciferase activity under the control of Foxp3 promoters upon KDM3A depletion further support that KDM3A regulates Foxp3 transcription by directly binding to its promoter. Despite these results that support the role of KDM3A in evading the immune system, further studies are needed to fully clarify its role in promoting lung adenocarcinoma growth.
As noted in breast cancers [29], KDM3A in lung cancer is also recruited to proliferation- and metastasis-related gene promoters by BRG1 [47]. BRG1 is pivotal for the expression of cyclin B1 and latent TGF binding protein 2 (LTBP2), the upregulation of which is correlated with malignant lung cancer growth. In lung cancer, hypoxia induces BRG1 to regulate the proliferation and migration of lung cancer cells by activating cyclin B1 and LTBP2, whereas loss of BRG1 via siRNA treatment decreases hypoxia-induced migration and proliferation. A series of ChIP assays indicated that hypoxia increases the interaction with and recruitment of KDM3A by BRG1 to proximal cyclin B1 and LTBP2 promoters. KDM3A recruitment to these promoters is decreased when BRG1 expression is abrogated, confirming that KDM3A interacts with BRG1 and controls the methylation status on these genes. Taken together, these data highlight the common role of KDM3A in different cancers, further reinforcing its prominent role in tumorigenesis.
2.5. Other Cancers
How KDM3A functions in tumorigenesis of other cancers can be found in this review. KDM3A induces proliferation and cancer growth in following cancers, but not limited to: liver cancer, bladder cancer, neuroblastoma, pancreatic cancer, ovarian cancer, and multiple myeloma.
3. KDM3 Inhibitors as Emerging Epigenetic Cancer Agents
Deregulation of KDM3 catalytic activity and expression is increasingly associated with the progression of various cancers. Indeed, extensive histone methylation modifications imply chemoresistance [28][48], patient relapse [20][21][26][31][34][49], and poor prognosis [20][21][44]. KDM3 proteins, particularly KDM3A, can act as oncoproteins, making them potential therapeutic targets for cancer treatment. KDM inhibitors can be categorized into four classes based on their catalytic mechanisms [50]; α-KG/2-OG cofactor mimics, metal cofactor disruptors, competitive substrate inhibitors, and substrate- and cofactor-independent inhibitors. The development of potent selective inhibitors for KDM is going through an active investigation, but only a few KDM3 inhibitors have been reported and they are pan-KDM inhibitors that are not selective for KDM3 subfamily. Recently, a few KDM3 modulators were reported as KDM3 JmjC domain binding molecules (Figure 3).
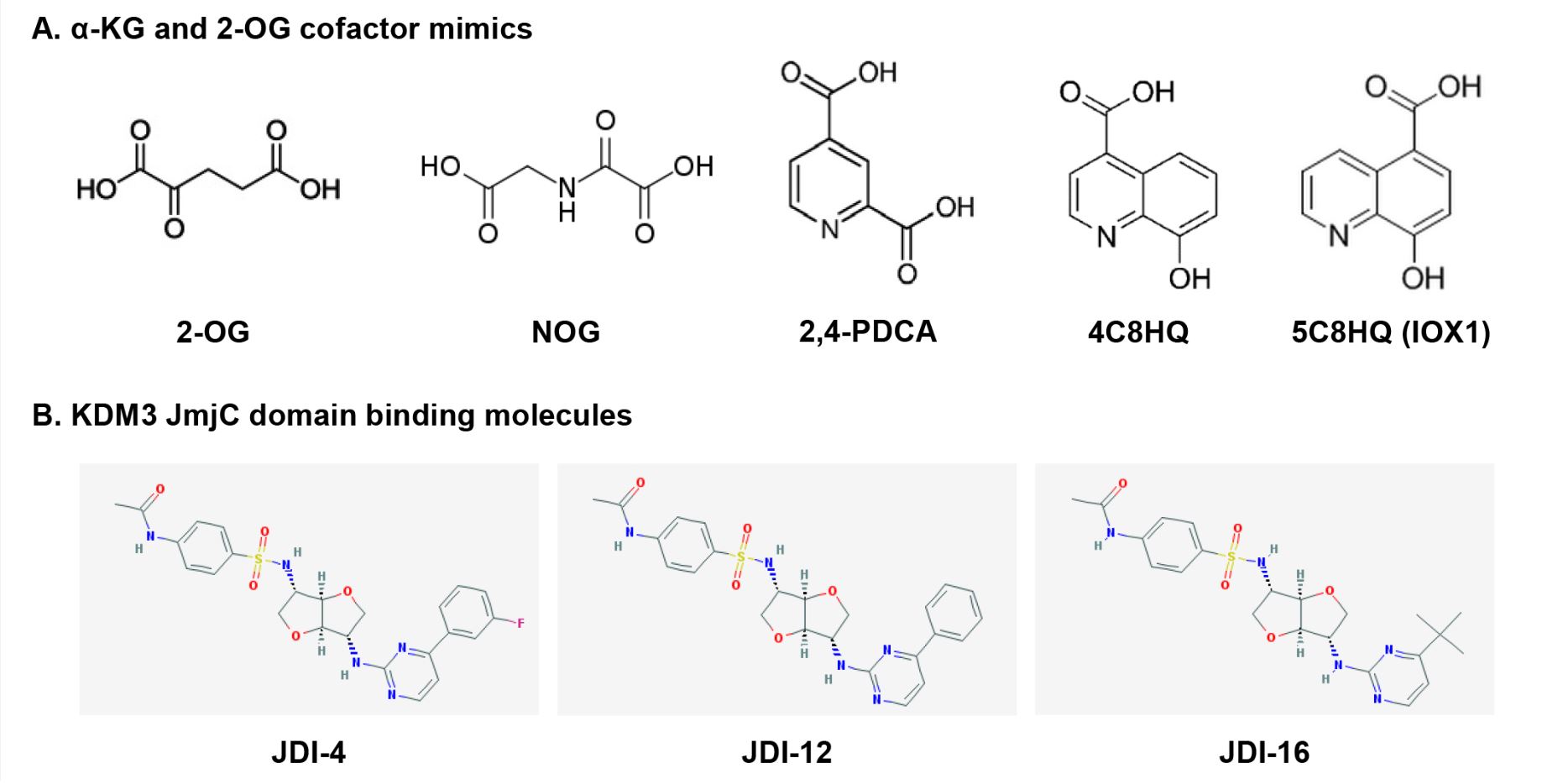
Figure 3. Chemical structures of KDM3 inhibitors and modulators: (a) cofactor mimics; (b) KDM3
JmjC domain-binding molecules.
3.1. α-KG/2-OG Cofactor Mimics: Metal-chelated Inhibitors
α-KG/2-OG is required for the demethylation of all JmjC-KDMs [4][6][8]. Most KDM inhibitors are of the first class that includes broad-spectrum α-KG/2-OG cofactor inhibitors. These inhibitors competitively bind Fe(II) (ferrous iron) molecules at the active catalytic site and inhibit all major regions of the JmjC-KDM proteins through the enzymes’ conserved Fe(II) and α-KG/2-OG sites [51][52]. Most α-KG/2-OG cofactor inhibitors are α-KG/2-OG competitors, including the oxayly acid-derivative N-oxalylglycine (NOG) [53], 2,4-pyridinedicarboxylates (2,4-PDCA) [37][54][55][56], hydroxamate [54], and hydroxyquinoline (HQ) derivatives [57] (Figure 3A). NOG was first identified as a 2-OG oxygenase inhibitor, and it is a closely related analog of α-KG/2-OG [58]. Hopkinson et al. investigated the comparative inhibition profiles for NOG, 2,4-PDCA, 5-carboxy-8-hydroxyquinoline (IOX1), and 4-carboxy-8-hydroxyquinoline (4C8HQ) for 14 active 2-OG oxygenases, including KDM3A [57], by in vitro assays. All four of the tested compounds inhibited every 2-OG oxygenases. However, the degree of inhibition for KDM3A varies substantially across the four inhibitors. Among the four inhibitors tested, IOX1 was the most potent inhibitor as it displayed IC50 values at 1 mM or lower against the KDM3A. IOX1 causes iron translocation in protein’s active-site metal position in respective to the position when the inhibitor is not present. This iron translocation may contribute to the variance in the efficiency of IOX1 and 4C8HQ, a regioisomer of IOX1 [59]. Although α-KG/2-OG cofactor analogs have proven to be broad-spectrum inhibitors, these compounds apparently lack the selectivity for each 2-OG oxygenase, including KDM and have poor cell penetration because of their structures.
3.2. Metal Cofactor Disruptor
KDM’s catalytic activity can be inhibited by interfering with iron cofactor binding using metal cofactor disruptors such as non-iron metals and organic molecules. Non-iron metals including nickel inhibit the catalytic activity of KDM3A by replacing the Fe(II) ion in the iron-binding pocket at the catalytic site to block the hydroxylation process [60]. The IC50 of nickel ions for KDM3A is 25 μM. However, many proteins contain nickel ion-binding sites and thus, the selectivity of iron-ejecting compounds for KDM3A requires further study.
3.3. KDM3 Modulators: KDM3 JmjC Domain-binding Molecules
Recently, Xu et al. reported new KDM3 modulators from a library of 149,519 natural products and 33,765 Chinese medicine components via virtual screening using the JmjC domain [61], which is a potential functional domain of KDM3 [10]. Among these compounds, JMJD1C Jumonji domain inhibitor 4 (JDI-4), JDI-12, and JDI-16 were identified via surface plasmon resonance analysis as potential KDM3B and KDM3C JmjC domain-binding molecules (Figure 3B). In vitro demethylation assays revealed that JDI-4 reverses H3K9me1 demethylation catalyzed by recombinant KDM3B, which is 57% similar to the JmjC domain of KDM3C. In vivo demethylation assays illustrated that JDI-4 and JDI-12 globally increase H3K9me1 levels. However, Xu et al. did not test the effect of JDIs on the inhibitory activity of other KDMs, including KDM3A. To draw conclusions regarding the biological activity of new inhibitors, it needs to be confirmed that their observed cellular effects are attributable to the inhibition of each KDM3 enzyme rather than non-specific effects or off-target activity, thus whether JDIs can function as novel KDM3-selective inhibitors requires further examination.
4. Conclusion
The latest research in epigenetics and epigenomics uncovered that epigenetic dysregulation is one of the essential causes of tumorigenesis. DNA methylation inhibitors have already been used for cancer treatment [63][64]. In addition to DNA methylation, interest in abnormal histone methylation and its implication in cancer has been growing. Given the number of links of KDM3A to tumorigenesis and metastasis, KDM3A stands as arising promising therapeutic target in cancer treatment. Indeed, the structure of some JmjC catalytic domains reveals that they are druggable because they are suitable to embrace small-molecule inhibitors [6][61][65]. Pan-KDM inhibitors have been invented through rational drug design and been found in compound screens (e.g., JIB-04) [37]. Despite the increasing number of KDM inhibitors in development, only a few drugs have produced satisfactory results and exhibited sufficient specificity for consideration as principal structures for drug development. Consequently, no currently reported compounds are selective for KDM3 subfamily members or their isoforms. Several reasons challenge the development of KDM3-selective inhibitors.
First, most, if not all, KDM3 crystal structures are undetermined. The lack of a critical residue in the Zn finger domain that specifically recognizes histone substrates at KDM3A makes it more difficult to develop specific inhibitors for KDM3A [10][61]. In contrast to the paucity of structural information regarding KDM3, structural information on KDM4A [6][65], KDM4D [66], KDM5A [67], KDM5B [68], KDM6A [69], and KDM8 [70] has been reported and selective inhibitors based on their structures have been developed [7][61][68][69][70]. Thus, the structure of KDM3 proteins needs to be elucidated to better understand its molecular mechanisms and facilitate the development of potent, and selective KDM3 inhibitors.
Second, most inhibitors are cofactors or substrate mimics with poorly understood mechanisms of action for specific KDM3s. Also, other inhibitors identified to date target the broad catalytic machinery of KDM and therefore have low specificity for KDM3. Thus, more research is needed to understand the biochemical selectivity of KDM3 to develop specific inhibitors. It is currently known that KDM3A homodimerizes through an association of its catalytic domains and demethylates H3K9 via a two-step process. The two active sites of the homodimer are pivotal for its enzymatic activity [14]. This catalytic mechanism is unlike that of other H3K9 demethylases such as KDM7B/PHF8 and KIAA1718 [71]. These findings hint at the growing possibility of developing small molecules that specifically inhibit KDM3A. Third, no conclusive data have validated the efficacy of KDM inhibitors against specific cancer types. These issues have prevented KDM inhibitors from progressing beyond basic research to clinical research and medical applications. Consequently, further studies are essential to discover promising inhibitors that can selectively inhibit specific KDMs including KDM3A, in vision of treating diseases not limited to cancers.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12051098
References
- Smith, B.C.; Denu, J.M.; Chemical mechanisms of histone lysine and arginine modifications. Acta 2009, 1789, 45-57, .
- Debasis Patnaik; Hang Gyeong Chin; Pierre-Olivier Estève; Jack Benner; Steven E. Jacobsen; Sriharsa Pradhan; Substrate Specificity and Kinetic Mechanism of Mammalian G9a Histone H3 Methyltransferase. Journal of Biological Chemistry 2004, 279, 53248-53258, 10.1074/jbc.m409604200.
- Woon Ki Paik; Sangduk Kim; Enzymatic demethylation of calf thymus histones. Biochemical and Biophysical Research Communications 1973, 51, 781-788, 10.1016/0006-291x(73)91383-1.
- Yu-Ichi Tsukada; Jia Fang; Hediye Erdjument-Bromage; Maria E. Warren; Christoph H. Borchers; Paul Tempst; Yi Zhang; Histone demethylation by a family of JmjC domain-containing proteins. Nature 2005, 439, 811-816, 10.1038/nature04433.
- Yujiang Shi; Fei Lan; Caitlin Matson; Peter Mulligan; Johnathan R. Whetstine; Philip A. Cole; Robert A. Casero; Yang Shi; Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941-953, 10.1016/j.cell.2004.12.012.
- Zhongzhou Chen; Jianye Zang; Johnathan Whetstine; Xia Hong; Foteini Davrazou; Tatiana G. Kutateladze; Michael Simpson; Qilong Mao; Cheol-Ho Pan; Shaodong Dai; et al. Structural Insights into Histone Demethylation by JMJD2 Family Members. Cell 2006, 125, 691-702, 10.1016/j.cell.2006.04.024.
- Dong Hoon Lee; Go Woon Kim; Yu Hyun Jeon; Jung Yoo; Sang Wu Lee; So Hee Kwon; Advances in histone demethylase KDM4 as cancer therapeutic targets. The FASEB Journal 2020, 34, 3461-3484, 10.1096/fj.201902584r.
- Johnathan R. Whetstine; Amanda Nottke; Fei Lan; Maite Huarte; Sarit Smolikov; Zhongzhou Chen; Eric Spooner; En Li; Gongyi Zhang; Monica Colaiacovo; et al. Reversal of Histone Lysine Trimethylation by the JMJD2 Family of Histone Demethylases. Cell 2006, 125, 467-481, 10.1016/j.cell.2006.03.028.
- Susanne Marije Kooistra; Kristian Helin; Molecular mechanisms and potential functions of histone demethylases. Nature Reviews Molecular Cell Biology 2012, 13, 297-311, 10.1038/nrm3327.
- Brauchle, M.; Yao, Z.; Arora, R. et al.; Protein complex interactor analysis and di erential activity of KDM3 subfamily members towards H3K9 methylation.. PLos One 2013, 8, e60549, .
- Kenichi Yamane; Charalambos Toumazou; Yu-Ichi Tsukada; Hediye Erdjument-Bromage; Paul Tempst; Jiemin Wong; Yi Zhang; JHDM2A, a JmjC-Containing H3K9 Demethylase, Facilitates Transcription Activation by Androgen Receptor. Cell 2006, 125, 483-495, 10.1016/j.cell.2006.03.027.
- Liang Liu; Hyunmi Kim; Alex Casta; Yuki Kobayashi; Lawrence S. Shapiro; Angela M. Christiano; Hairless is a histone H3K9 demethylase. The FASEB Journal 2013, 28, 1534-1542, 10.1096/fj.13-237677.
- Robert J. Klose; Eric M. Kallin; Yi Zhang; JmjC-domain-containing proteins and histone demethylation. Nature Reviews Genetics 2006, 7, 715-727, 10.1038/nrg1945.
- Satoshi Goda; Takayuki Isagawa; Yoko Chikaoka; Takeshi Kawamura; Hiroyuki Aburatani; Control of Histone H3 Lysine 9 (H3K9) Methylation State via Cooperative Two-step Demethylation by Jumonji Domain Containing 1A (JMJD1A) Homodimer*. Journal of Biological Chemistry 2013, 288, 36948-36956, 10.1074/jbc.M113.492595.
- Christer Höög; Martin Schalling; Bertil Daneholt; Eva Grunder-Brundell; Analysis of a murine male germ cell-specific transcript that encodes a putative zinc finger protein. Molecular Reproduction and Development 1991, 30, 173-181, 10.1002/mrd.1080300302.
- Yuki Okada; Greg Scott; Manas K. Ray; Yuji Mishina; Yi Zhang; Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature 2007, 450, 119-123, 10.1038/nature06236.
- Keisuke Tateishi; Yuki Okada; Eric M. Kallin; Yi Zhang; Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 2009, 458, 757-761, 10.1038/nature07777.
- Yuin-Han Loh; Weiwei Zhang; X. Chen; Joshy George; Huck-Hui Ng; Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes & Development 2007, 21, 2545-2557, 10.1101/gad.1588207.
- Shunsuke Kuroki; Shogo Matoba; Mika Akiyoshi; Yasuko Matsumura; Hitoshi Miyachi; Nathan Mise; Kuniya Abe; Atsuo Ogura; Dagmar Wilhelm; Peter Koopman; et al. Epigenetic Regulation of Mouse Sex Determination by the Histone Demethylase Jmjd1a. Science 2013, 341, 1106-1109, 10.1126/science.1239864.
- Lingling Fan; Guihong Peng; Natasha Sahgal; Ladan Fazli; Martin Gleave; Yuji Zhang; Arif Hussain; Jianfei Qi; Regulation of c-Myc expression by the histone demethylase JMJD1A is essential for prostate cancer cell growth and survival. Oncogene 2015, 35, 2441-2452, 10.1038/onc.2015.309.
- Xiaolu Cui; Chiyuan Piao; Chengcheng Lv; Xuyong Lin; Zhe Zhang; Xiankui Liu; ZNFX1 anti-sense RNA 1 promotes the tumorigenesis of prostate cancer by regulating c-Myc expression via a regulatory network of competing endogenous RNAs. Cellular and Molecular Life Sciences 2019, 77, 1135-1152, 10.1007/s00018-019-03226-x.
- Hyun-Soo Cho; Gouji Toyokawa; Yataro Daigo; Shinya Hayami; Ken Masuda; Noriko Ikawa; Yuka Yamane; Kazuhiro Maejima; Tatsuhiko Tsunoda; Helen I. Field; et al. The JmjC domain-containing histone demethylase KDM3A is a positive regulator of the G1/S transition in cancer cells via transcriptional regulation of the HOXA1 gene. International Journal of Cancer 2011, 131, E179-E189, 10.1002/ijc.26501.
- Prasad Dandawate; Chandrayee Ghosh; Kanagaraj Palaniyandi; Santanu Paul; Sonia Rawal; Rohan Pradhan; Afreen Asif Ali Sayed; Sonali Choudhury; David Standing; Dharmalingam Subramaniam; et al. The Histone Demethylase KDM3A, Increased in Human Pancreatic Tumors, Regulates Expression of DCLK1 and Promotes Tumorigenesis in Mice.. Gastroenterology 2019, 157, 1646-1659.e11, 10.1053/j.gastro.2019.08.018.
- Ho-Youl Lee; Eun Gyeong Yang; Hyunsung Park; Hypoxia enhances the expression of prostate-specific antigen by modifying the quantity and catalytic activity of Jumonji C domain-containing histone demethylases. Carcinogenesis 2013, 34, 2706-2715, 10.1093/carcin/bgt256.
- Maina, P.K.; Shao, P.; Liu, Q.; Fazli, L.; Tyler, S.; Nasir, M.; Dong, X.; Qi, H.H.; c-MYC drives histone demethylase PHF8 during neuroendocrine di erentiation and in castration-resistant prostate cancer. Oncotarget 2016, 7, 75585-75602, .
- Peng, K.; Su, G.; Ji, J.; Yang, X.; Miao, M.; Mo, P.; Li, M.; Xu, J.; Li,W.; Yu, C.; et al. Histone demethylase JMJD1A promotes colorectal cancer growth and metastasis by enhancing Wnt/beta-catenin signaling.. J. Biol. Chem. 2018, 293, 10606-10619, .
- Zhe Wang; Xin Yang; Cheng Liu; Xin Li; Buyu Zhang; Bo Wang; Yu Zhang; Chen Song; Tianzhuo Zhang; Minghui Liu; et al. Acetylation of PHF5A Modulates Stress Responses and Colorectal Carcinogenesis through Alternative Splicing-Mediated Upregulation of KDM3A. Molecular Cell 2019, 74, 1250-1263.e6, 10.1016/j.molcel.2019.04.009.
- Sivakumar Ramadoss; Gao Guo; Cun-Yu Wang; Lysine demethylase KDM3A regulates breast cancer cell invasion and apoptosis by targeting histone and the non-histone protein p53. Oncogene 2016, 36, 47-59, 10.1038/onc.2016.174.
- Lina Sun; Yibiao Yuan; Junliang Chen; Changlan Ma; Yong Xu; Brahma related gene 1 (BRG1) regulates breast cancer cell migration and invasion by activating MUC1 transcription. Biochemical and Biophysical Research Communications 2019, 511, 536-543, 10.1016/j.bbrc.2019.02.088.
- Stephen Wilson; Lingling Fan; Natasha Sahgal; Jianfei Qi; Fabian V. Filipp; The histone demethylase KDM3A regulates the transcriptional program of the androgen receptor in prostate cancer cells. Oncotarget 2017, 8, 30328-30343, 10.18632/oncotarget.15681.
- Mahajan, K.; Lawrence, H.R.; Lawrence, N.J.; Mahajan, N.P. ACK1 tyrosine kinase interacts with histone demethylase KDM3A to regulate the mammary tumor oncogene HOXA1. J. Biol. Chem. 2014, 289, 28179–28191.
- Wade, M.A.; Jones, D.;Wilson, L.; Stockley, J.; Co ey, K.; Robson, C.N.; Gaughan, L. The histone demethylase enzyme KDM3A is a key estrogen receptor regulator in breast cancer. Nucleic Acids Res. 2015, 43, 196–207.
- Jones, D.;Wilson, L.; Thomas, H.; Gaughan, L.;Wade, M.A. The Histone Demethylase Enzymes KDM3A and KDM4B Co-Operatively Regulate Chromatin Transactions of the Estrogen Receptor in Breast Cancer. Cancers (Basel) 2019, 11, 1122.
- Li, J.; Yu, B.; Deng, P.; Cheng, Y.; Yu, Y.; Kevork, K.; Ramadoss, S.; Ding, X.; Li, X.; Wang, C.Y. KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/beta-catenin signalling. Nat. Commun. 2017, 8, 15146.
- Bjorkman, M.; Ostling, P.; Harma, V.; Virtanen, J.; Mpindi, J.P.; Rantala, J.; Mirtti, T.; Vesterinen, T.; Lundin, M.; Sankila, A.; et al. Systematic knockdown of epigenetic enzymes identifies a novel histone demethylase PHF8 overexpressed in prostate cancer with an impact on cell proliferation, migration and invasion. Oncogene 2012, 31, 3444–3456.
- Fan, L.; Zhang, F.; Xu, S.; Cui, X.; Hussain, A.; Fazli, L.; Gleave, M.; Dong, X.; Qi, J. Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E4584–E4593.
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A.M.; Ruiz, J.; Bruick, R.; Pena-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 2035.
- Howard, J.H.; Bland, K.I. Current management and treatment strategies for breast cancer. Curr. Opin. Obstet. Gynecol. 2012, 24, 44–48.
- Zhao, Q.Y.; Lei, P.J.; Zhang, X.; Zheng, J.Y.; Wang, H.Y.; Zhao, J.; Li, Y.M.; Ye, M.; Li, L.; Wei, G.; et al. Global histone modification profiling reveals the epigenomic dynamics during malignant transformation in a four-stage breast cancer model. Clin. Epigenet. 2016, 8, 34.
- Li, X.; Oh, S.; Song, H.; Shin, S.; Zhang, B.; Freeman, W.M.; Janknecht, R. A potential common role of the Jumonji C domain-containing 1A histone demethylase and chromatin remodeler ATRX in promoting colon cancer. Oncol. Lett. 2018, 16, 6652–6662.
- Liu, J.; Liang, T.; Zhangsun, W. KDM3A is associated with tumor metastasis and modulates colorectal cancer cell migration and invasion. Int. J. Biol. Macromol. 2019, 126, 318–325.
- Maia, L.L.; Peterle, G.T.; Dos Santos, M.; Trivilin, L.O.; Mendes, S.O.; de Oliveira, M.M.; Dos Santos, J.G.; Stur, E.; Agostini, L.P.; Couto, C.; et al. JMJD1A, H3K9me1, H3K9me2 and ADM expression as prognostic markers in oral and oropharyngeal squamous cell carcinoma. PLoS ONE 2018, 13, e0194884.
- Krieg, A.J.; Rankin, E.B.; Chan, D.; Razorenova, O.; Fernandez, S.; Giaccia, A.J. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol. Cell. Biol. 2010, 30, 344–353.
- Wang, H.Y.; Long, Q.Y.; Tang, S.B.; Xiao, Q.; Gao, C.; Zhao, Q.Y.; Li, Q.L.; Ye, M.; Zhang, L.; Li, L.Y.; et al. Histone demethylase KDM3A is required for enhancer activation of hippo target genes in colorectal cancer. Nucleic Acids Res. 2019, 47, 2349–2364.
- Zuo, X.; Morris, J.S.; Shureiqi, I. Chromatin modification requirements for 15-lipoxygenase-1 transcriptional reactivation in colon cancer cells. J. Biol. Chem. 2008, 283, 31341–31347.
- Li, Y.; Yang, W.; Wu, B.; Liu, Y.; Li, D.; Guo, Y.; Fu, H.; Li, Y. KDM3A promotes inhibitory cytokines secretion by participating in TLR4 regulation of Foxp3 transcription in lung adenocarcinoma cells. Oncol. Lett. 2017, 13, 3529–3537.
- Li, Z.; Xia, J.; Fang, M.; Xu, Y. Epigenetic regulation of lung cancer cell proliferation and migration by the chromatin remodeling protein BRG1. Oncogenesis 2019, 8, 66.
- Ramadoss, S.; Sen, S.; Ramachandran, I.; Roy, S.; Chaudhuri, G.; Farias-Eisner, R. Lysine-specific demethylase KDM3A regulates ovarian cancer stemness and chemoresistance. Oncogene 2017, 36, 1537–1545.
- Yamada, D.; Kobayashi, S.; Yamamoto, H.; Tomimaru, Y.; Noda, T.; Uemura, M.;Wada, H.; Marubashi, S.; Eguchi, H.; Tanemura, M.; et al. Role of the hypoxia-related gene, JMJD1A, in hepatocellular carcinoma: Clinical impact on recurrence after hepatic resection. Ann. Surg. Oncol. 2012, 19 (Suppl. 3), S355–S364.
- Chin, Y.W.; Han, S.Y. KDM4 histone demethylase inhibitors for anti-cancer agents: A patent review. Expert Opin. Ther. Pat. 2015, 25, 135–144.
- McDonough, M.A.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672.
- Itoh, Y.; Suzuki, T.; Miyata, N. Small-molecular modulators of cancer-associated epigenetic mechanisms. Mol. Biosyst. 2013, 9, 873–896.
- Cloos, P.A.; Christensen, J.; Agger, K.; Maiolica, A.; Rappsilber, J.; Antal, T.; Hansen, K.H.; Helin, K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006, 442, 307–311.
- Rose, N.R.; Ng, S.S.; Mecinovic, J.; Lienard, B.M.; Bello, S.H.; Sun, Z.; McDonough, M.A.; Oppermann, U.; Schofield, C.J. Inhibitor sca olds for 2-oxoglutarate-dependent histone lysine demethylases. J. Med. Chem. 2008, 51, 7053–7056.
- Sekirnik, R.; Rose, N.R.; Thalhammer, A.; Seden, P.T.; Mecinovic, J.; Schofield, C.J. Inhibition of the histone lysine demethylase JMJD2A by ejection of structural Zn(II). Chem. Commun. (Camb) 2009, 42, 6376–6378.
- Thalhammer, A.; Mecinovic, J.; Loenarz, C.; Tumber, A.; Rose, N.R.; Heightman, T.D.; Schofield, C.J. Inhibition of the histone demethylase JMJD2E by 3-substituted pyridine 2,4-dicarboxylates. Org. Biomol. Chem. 2011, 9, 127–135.
- Hopkinson, R.J.; Tumber, A.; Yapp, C.; Chowdhury, R.; Aik, W.; Che, K.H.; Li, X.S.; Kristensen, J.B.L.; King, O.N.F.; Chan, M.C.; et al. 5-Carboxy-8-hydroxyquinoline is a Broad Spectrum 2-Oxoglutarate Oxygenase Inhibitor which Causes Iron Translocation. Chem. Sci. 2013, 4, 3110–3117.
- Cunli e, C.J.; Franklin, T.J.; Hales, N.J.; Hill, G.B. Novel inhibitors of prolyl 4-hydroxylase. 3. Inhibition by the substrate analogue N-oxaloglycine and its derivatives. J. Med. Chem. 1992, 35, 2652–2658.
- Thinnes, C.C.; England, K.S.; Kawamura, A.; Chowdhury, R.; Schofield, C.J.; Hopkinson, R.J. Targeting histone lysine demethylases—Progress, challenges, and the future. Biochim. Biophys. Acta 2014, 1839, 1416–1432.
- Chen, H.; Kluz, T.; Zhang, R.; Costa, M. Hypoxia and nickel inhibit histone demethylase JMJD1A and repress Spry2 expression in human bronchial epithelial BEAS-2B cells. Carcinogenesis 2010, 31, 2136–2144.
- Xu, X.;Wang, L.; Hu, L.; Dirks,W.G.; Zhao, Y.;Wei, Z.; Chen, D.; Li, Z.;Wang, Z.; Han, Y.; et al. Small molecular modulators of JMJD1C preferentially inhibit growth of leukemia cells. Int. J. Cancer 2020, 146, 400–412.
- Shi, Y.; Whetstine, J.R. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell 2007, 25, 1–14.
- Rodriguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339.
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727.
- Ng, S.S.; Kavanagh, K.L.; McDonough, M.A.; Butler, D.; Pilka, E.S.; Lienard, B.M.; Bray, J.E.; Savitsky, P.; Gileadi, O.; von Delft, F.; et al. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature 2007, 448, 87–91.
- Krishnan, S.; Trievel, R.C. Structural and functional analysis of JMJD2D reveals molecular basis for site-specific demethylation among JMJD2 demethylases. Structure 2013, 21, 98–108.
- Horton, J.R.; Liu, X.; Gale, M.; Wu, L.; Shanks, J.R.; Zhang, X.; Webber, P.J.; Bell, J.S.K.; Kales, S.C.; Mott, B.T.; et al. Structural Basis for KDM5A Histone Lysine Demethylase Inhibition by Diverse Compounds. Cell Chem. Biol. 2016, 23, 769–781.
- Johansson, C.; Velupillai, S.; Tumber, A.; Szykowska, A.; Hookway, E.S.; Nowak, R.P.; Strain-Damerell, C.; Gileadi, C.; Philpott, M.; Burgess-Brown, N.; et al. Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat. Chem. Biol. 2016, 12, 539–545.
- Sengoku, T.; Yokoyama, S. Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes Dev. 2011, 25, 2266–2277.
- Wang, H.; Zhou, X.; Wu, M.; Wang, C.; Zhang, X.; Tao, Y.; Chen, N.; Zang, J. Structure of the JmjC-domain-containing protein JMJD5. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1911–1920.
- Horton, J.R.; Upadhyay, A.K.; Qi, H.H.; Zhang, X.; Shi, Y.; Cheng, X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 2010, 17, 38–43.
- Horton, J.R.; Upadhyay, A.K.; Qi, H.H.; Zhang, X.; Shi, Y.; Cheng, X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 2010, 17, 38–43.
- Horton, J.R.; Upadhyay, A.K.; Qi, H.H.; Zhang, X.; Shi, Y.; Cheng, X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 2010, 17, 38–43.