Phosphoribosyl pyrophosphate synthetase (PRS EC 2.7.6.1) is a rate-limiting enzyme that irreversibly catalyzes the formation of phosphoribosyl pyrophosphate (PRPP) from ribose-5-phosphate and adenosine triphosphate (ATP). This key metabolite is required for the synthesis of purine and pyrimidine nucleotides, the two aromatic amino acids histidine and tryptophan, the cofactors nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+), all of which are essential for various life processes. Despite its ubiquity and essential nature across the plant and animal kingdoms, PRPP synthetase displays species-specific characteristics regarding the number of gene copies and architecture permitting interaction with other areas of cellular metabolism. The impact of mutated PRS genes in the model eukaryote Saccharomyces cerevisiae on cell signalling and metabolism may be relevant to the human neuropathies associated with PRPS mutations. Human PRPS1 and PRPS2 gene products are implicated in drug resistance associated with recurrent acute lymphoblastic leukaemia and progression of colorectal cancer and hepatocellular carcinoma.
- phosphoribosyl pyrophosphate synthetase (PRS/PRPS)
- eukaryotic model organisms
- Saccharomyces cerevisiae
- Danio rerio
- human neuropathies: Arts syndrome
- CMTX5
- DFN2/DFNX1
- ageing
- cancer
- cell signalling
- CWI pathway
1. Introduction
Phosphoribosyl pyrophosphate synthetase (PRS; ATP:D-ribose-5-phosphate pyrophosphotransferase, EC 2.7.6.1) is an essential rate-limiting enzyme that catalyzes the irreversible transformation of ribose-5-phosphate in the presence of ATP to produce 5-phospho-D-ribosyl-α-1-pyrophosphate (PRPP) and AMP and is subjected to feedback inhibition by AMP, ADP and GDP [1][2][3][4][5]. PRS links carbon and nitrogen in cellular metabolism producing the high-energy compound PRPP, a key metabolite necessary for the biosynthesis of purine and pyrimidine nucleotides, the cofactors nicotinamide adenine dinucleotide (NAD+), nicotinamide adenine dinucleotide phosphate (NADP+) and, in specific organisms, e.g., Escherichia coli, the amino acids histidine and tryptophan [6][7]. PRS is responsible not only for the de novo synthesis of purine nucleotides but also plays a role in the salvage pathways via dedicated phosphoribosyl transferases for adenine, guanine and hypoxanthine. However, pyrimidine nucleotides, i.e., thymine and uridine, are salvaged by recycling from degradation products of DNA and RNA [8][9][10][11].
Synthesis of PRPP is essential for all free-living organisms but is lacking in parasites, e.g., Trypanosoma and Leishmania, which are dependent on the host metabolism for the synthesis of PRPP [12]. PRS enzymes have been identified in numerous organisms including mammals, human and rats [3][13][14][15][16][17]; plants, including Spinacia oleracea [18] and Arabidopsis thaliana [19][20]; bacteria, including Bacillus subtilis [21], and Bacillus pseudomallei [22]; yeasts, including Saccharomyces cerevisiae [23][24][25][26] and Schizosaccharomyces pombe [27]; and more recently, in zebrafish Danio rerio [28][29]; confirming the ubiquity of PRS genes across the five plant and animal kingdoms. There is now a plethora of PRS genes available for investigating the ramifications of altered PRPP synthetase on the physiology of various organisms (Figure 1).
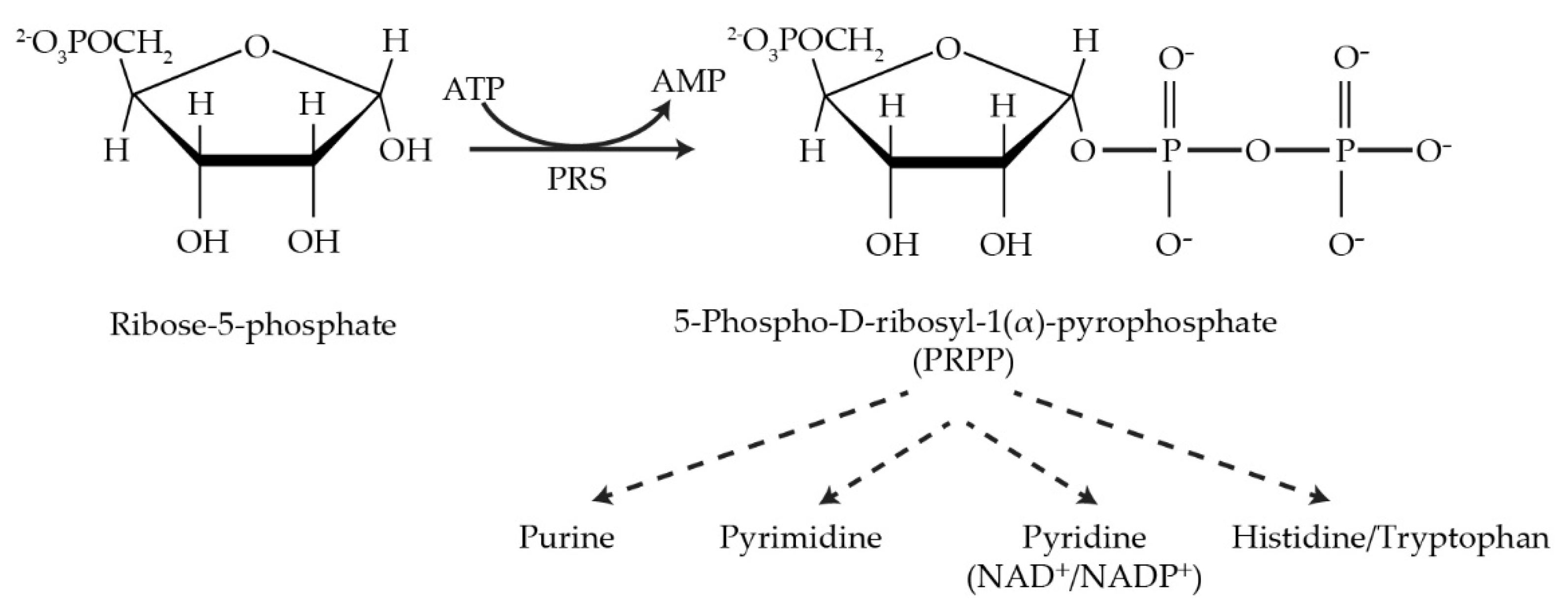
Figure 1. The reaction catalyzed by PRPP synthetase and metabolic pathways utilizing its product, PRPP.
Interestingly, the number of PRS genes in the genome varies from organism to organism. For instance, humans (HGNC PRPS), S. pombe (PomBase) and Aspergillus nidulans (AspGD) each possess three PRS genes, whereas S. oleracea (Spinach Base) and Eremothecium (Ashbya gossypii (Ashbya Genome Database)) genomes each contain four PRS genes. However, S. cerevisiae (Sacharomyces Genome Database (SGD)) and A. thaliana (AtGDB) harbour five PRS genes in their genomes. Irrespective of the number of PRS genes identified in different organisms, each organism must have at least a minimum of one gene coding for PRS to ensure survival, as is the case for bacteria [7][21][30]. An interesting discovery is that a gene, HbPRS4, identified in the genome of the rubber tree plant Hevea brasiliensis Műll, a member of the family Euphorbiacaea [31][32], encodes a protein with 80% identity to Prs4 of A. thaliana. A BlastN study revealed a second sequence in the rubber tree genome with 84% similarity to the HbPRS4 gene. The expression of both genes responded positively to ethylene treatment, as did the ATP/ADP content of latex cells, thus linking PRPP synthesis to natural rubber production [33][34]. In a similar context, it has been reported that in an industrial yeast strain, the increased expression of the genes required for the synthesis of PRPP correlated with enhanced xylose utilization, emphasizing the significant role of PRPP for biofuel production from lignocellulose [35][36]. More specifically, it has been shown that overexpression of ScPRS3 increases the resistance of the yeast cell wall to acetic acid stress, thus illustrating the importance of Prs3 in the enhancement of commercially important pathways, e.g., biofuel production for lignocellulose, possibly as a result of improving cell wall strength [37][38]. Mycobacterium tuberculosis, the pathogen responsible for tuberculosis, contains a single PRS gene whose product is the only source of PRPP for a precursor of the bacterial cell wall and therefore is essential for the maintenance of cell integrity, thus providing a potential drug target in the treatment of tuberculosis [39][40][41][42].
Numerous scientific reports have shown that mutations in the PRS genes in different organisms cause various forms of cellular disruption, including neurological diseases and metabolic disorders. The nomenclature for PRS genes is species-specific: the PRS genes in humans are designated as hPRPS, of which there are three isoforms, hPRPS1, hPRPS2 and hPRPS3 (hPRPS1L1). The genes, hPRPS1 and hPRSP2, are located on the long and short arms of the X-chromosome at positions Xq22.3 (OMIM 311850) and Xp22.2 (OMIM 311860) [43][44], respectively, whereas hPRPS3 maps to chromosome 7 at position p21.1 (OMIM 611566) and is expressed specifically in the testis [15][17][45]. Missense mutations in the hPRPS1 gene may lead to a gain-of-function associated with PRPS1 superactivity [46]. Other neurological disorders, such as Arts syndrome [47][48], syndromic and nonsyndromic sensorineural deafness (DFN2/DFNX-2) [49][50] and Charcot-Marie-Tooth disease type 5 (CMTX5) [51] are due to the loss-of-function in the hPRPS1 gene.
The sequence similarity of hPRPS proteins to those of the established eukaryotic model organisms S. cerevisiae (baker’s yeast) and D. rerio (zebrafish) has the potential to be instrumental in elucidating the ramifications of PRPP metabolism in human neuropathies. Biochemical and genetic studies have also identified the specific roles of hPRPS1 and hPRPS2 genes in one of the deadliest diseases, cancer. For instance, an hPRPS2 knockout in experimental models has resulted in c-Myc-driven tumorigenesis [52][53], suggesting that hPRPS2 may be a target protein for manipulating cancer cells, whereas it has been reported that a decrease in hPRPS1 expression impairs the proliferation of tumour cells [54]. Both hPRPS1 and hPRPS2 genes have been identified in pluripotent stem cells, where they may contribute to increased stem-cell-associated biosynthetic capacity [55].
2. PRS-Encoding Genes in Saccharomyces cerevisiae
S. cerevisiae, a workhorse eukaryotic model organism which, in addition to its contribution to bread, wine, beer and Marmite® production, has been exploited by biotechnologists for genetic and metabolic engineering by revamping and minimizing its genome through the ‘design-build-test-learn’ cycle of synthetic biology [56][57][58].
The whole yeast genome duplication that took place approximately 100 million years ago created a tetraploid yeast [59][60]. Over time, this unstable tetraploid yeast lost more than 80% of the duplicated gene copies. Nevertheless, of the remaining 6000 genes that make up the genome of the current species of S. cerevisiae, at least 10% are duplicated [61]. This implies that the remaining gene duplications must have made a positive contribution to the ancestral yeast’s relative fitness. Gene duplications provide a potential for increasing diversity, thereby permitting adaptation to new environments. If the products of the two gene copies are identical and autonomous, then one of the copies is available for alteration or can be lost. The alteration may provide a new link to the function of the other copy [59][62][63]. When the duplicated genes have interdependent functions, the chance of diversification is limited, but this can be overcome by retaining both the original and the altered copy since they are essential for the survival of the organism. Of the myriad gene products common to both humans and yeast, PRS in yeast is encoded by five unlinked paralogous genes as the result of genome duplication and subsequent evolution. The central role played by PRS gene products in cellular metabolism can be exploited to uncover potential therapeutic targets for PRPS-associated human diseases.
2.1. The Role of NHR1-1 in the Provision of PRPP and Maintenance of CWI
The S. cerevisiae genome contains five ScPRS genes, namely ScPRS1, ScPRS2, ScPRS3, ScPRS4 and ScPRS5, which are located on chromosomes XI, V, VIII, II, and XV, respectively [24][25][26]. Remarkably, ScPRS1 and ScPRS5 have acquired in-frame non-homologous regions (NHRs), which are not introns since they are still present in the mature polypeptides, as demonstrated by Western blotting with tailor-made antibodies [24][25][64]. The gene products Prs1 and Prs5 differ in length from Prs2 and Prs4 since they contain non-homologous regions NHR1-1, NHR5-1 and NHR5-2. In each instance, one of the component monomers of the essential minimal functional units, Prs1/Prs3, Prs2/Prs5 and Prs4/Prs5, contains an NHR. Both NHR1-1 with a length of 105 aa and NHR5-2 with 65 aa disrupt the catalytic flexible loops of Prs1 and Prs5 (Figure 2), respectively, rendering these polypeptides incapable of PRPP synthesis. NHR5-1 has a length of 113 aa and is located within the regulatory flexible loop and contains no less than nine predicted serine, threonine or tyrosine phosphorylation sites (PhosphoGrid and Phosphopep) [65][66]. A distinguishing feature of NHR1-1 and NHR5-1 is that they contain aa (amino acids) motifs associated with phosphorylation or ubiquitination [67]. S199 is located just prior to the beginning of the NHR1-1 sequence and has been postulated to play a role in mTORC1 signalling [68]. In the case of NHR5-1 S183, located centrally, it has been identified as being phosphorylated after rapamycin treatment [68], although there are other possible candidate aa residues in the near vicinity [69]. NHR5-2 is a stretch of 65 aa, including five serine residues, of which three, at the neighbouring positions S364, S367 and S369, are phosphorylatable [70]. Interestingly, characterization of a rapamycin-sensitive phosphoproteome revealed that Prs5 is hyperphosphorylated following rapamycin treatment [71][72], but the locations of the phosphosites were not specified. A high sequence similarity of 77% has been found for Prs1 between S. cerevisiae and A. gossypii (Ashbya Genome Database, AER083c) [73]. This similarity is maintained across the N-terminal sections of NHR1-1 in both species up to the midpoint of NHR1-1 of S. cerevisiae (unpublished data). Loss of the N-terminal portion of NHR1-1 in S. cerevisiae coincides with the inability to respond to heat shock as measured by Rlm1 expression, a transcriptional readout of the CWI (cell wall integrity) pathway [74]. NHR3-1 (Figure 2), close to the C-terminus of Prs3, is the pentameric sequence 284KKCPK288, corresponding to an NLS (nuclear localization signal) consensus sequence of two positively charged aa (K (lysine) or R (arginine)) flanking three residues, one of which is P (proline), as described in [75].
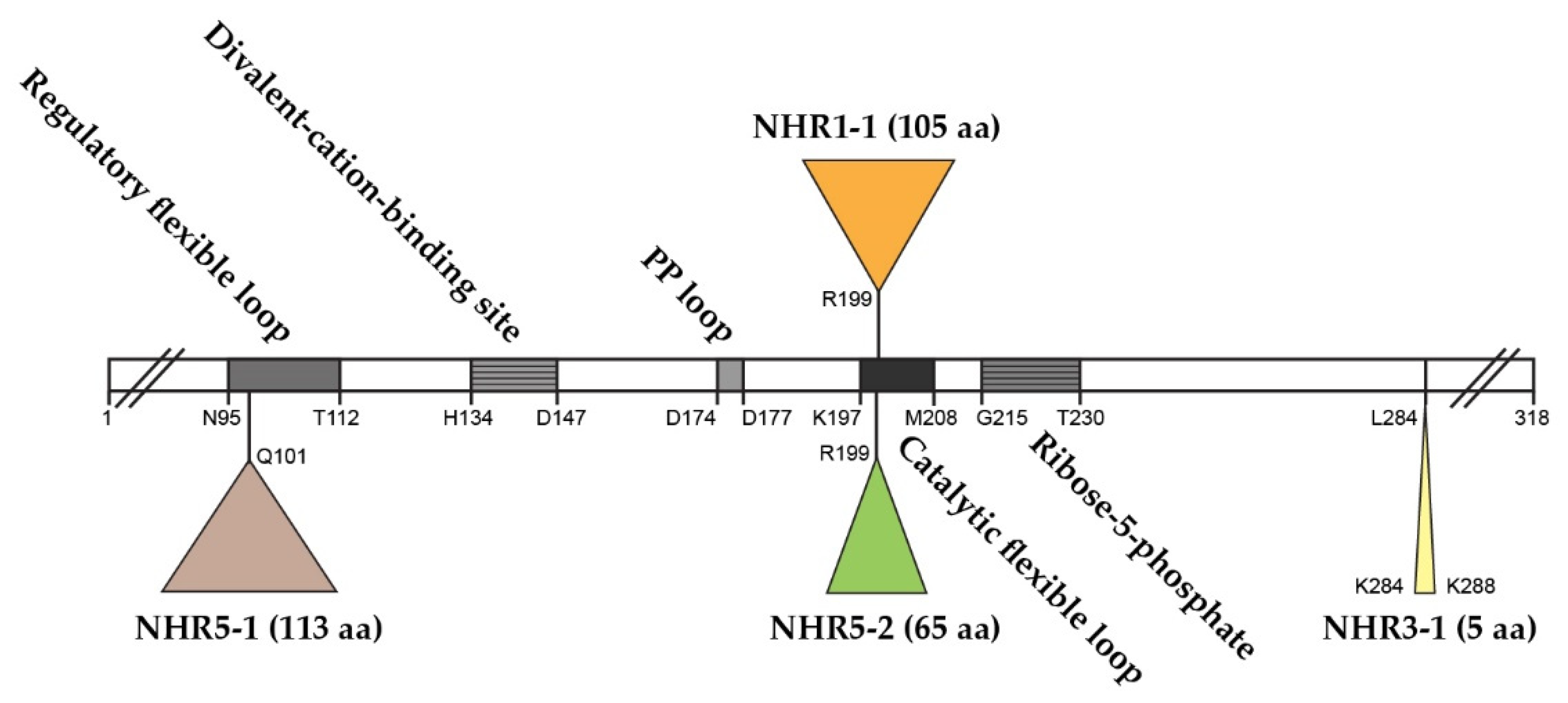
Figure 2. Relative positions of the non-homologous regions—NHR1-1, NHR3-1, NHR5-1 and NHR5-2—in the Prs2 polypeptide as the prototype. The Prs2 polypeptide consists of 318 aa and is indicated as an open bar with characteristic motifs therein. PP = pyrophosphate. The NHRs are positioned in Prs2 as triangles above or below the open bar. The coordinates of the four insertions reflect their lengths in Prs1, Prs3 and Prs5, respectively. The insertion points are defined where the similarity between Prs2 and Prs1 or Prs5 falls off. Sequence comparison was performed using the pairwise sequence alignment programme EMBOSS Needle.
A systematic phenotypic analysis revealed that individual deletion of any of the five S. cerevisiae PRS genes (ScPRS1-ScPRS5) did not seriously affect the cell’s viability [23][24][25][76]. However, S. cerevisiae cannot survive with a single PRS gene. Genetic evidence and Y2H (yeast-two-hybrid analysis) have shown that PRS exists as two interacting complexes: one as a heterodimer (Prs1/Prs3) and the other as a heterotrimer (Prs2/Prs4/Prs5). Furthermore, at least one of the three different combinatorial functional units, Prs1/Prs3, Prs2/Prs5 or Prs4/Prs5, must be present to ensure viability [26][76]. Expression of the five possible pairwise combinations of the ScPRS gene products in E. coli achieved only 25% of the level of the E. coli PRPP synthetase with only the combination, Prs1/Prs3 [77] which correlates with Prs1/Prs3 being the most important heterodimer for yeast survival [25]. Mutation of ScPRS1, ScPRS3 and ScPRS5 have proven useful for investigating the link between Prs and its specific functions—nucleotide biosynthesis, cell signalling and metabolism [26][76][78][79][80]. Overexpression of Pkc1 had a positive effect on the transcriptional activation of Rlm1 in either prs1Δ and prs3Δ strains at ambient and elevated temperatures and following α-factor stimulation [78].
A reduction in the capacity to synthesize PRPP, severe loss of enzymatic activity, compromised CWI, increased chitin content and severe growth retardation in S. cerevisiae were observed when ScPRS1/ScPRS3 were simultaneously deleted in the absence of ScPRS2 or ScPRS4 (Figure 3a). Therefore, the Prs1/Prs3 heterodimer is the most important [25][74] since the mutant strains relying on either Prs2/Prs5 or Prs4/Prs5 are severely impaired in their growth, their PRPP-synthesizing capacities and CWI, thus providing genetic evidence to support the corresponding enzyme activities measured in yeast. Furthermore, yeast PRS enzyme activity was reduced to 80% when ScPRS2, ScPRS4 and ScPRS5 were deleted simultaneously, but there was no influence on nucleotide content or growth rate (Figure 3b) [26][76][80]. Simultaneous deletion of ScPRS1 and ScPRS5 or ScPRS3 and ScPRS5 causes synthetic lethality, as does deletion of ScPRS2 and ScPRS4 in addition to the loss of ScPRS1 or ScPRS3 [26][76] (Figure 3c).
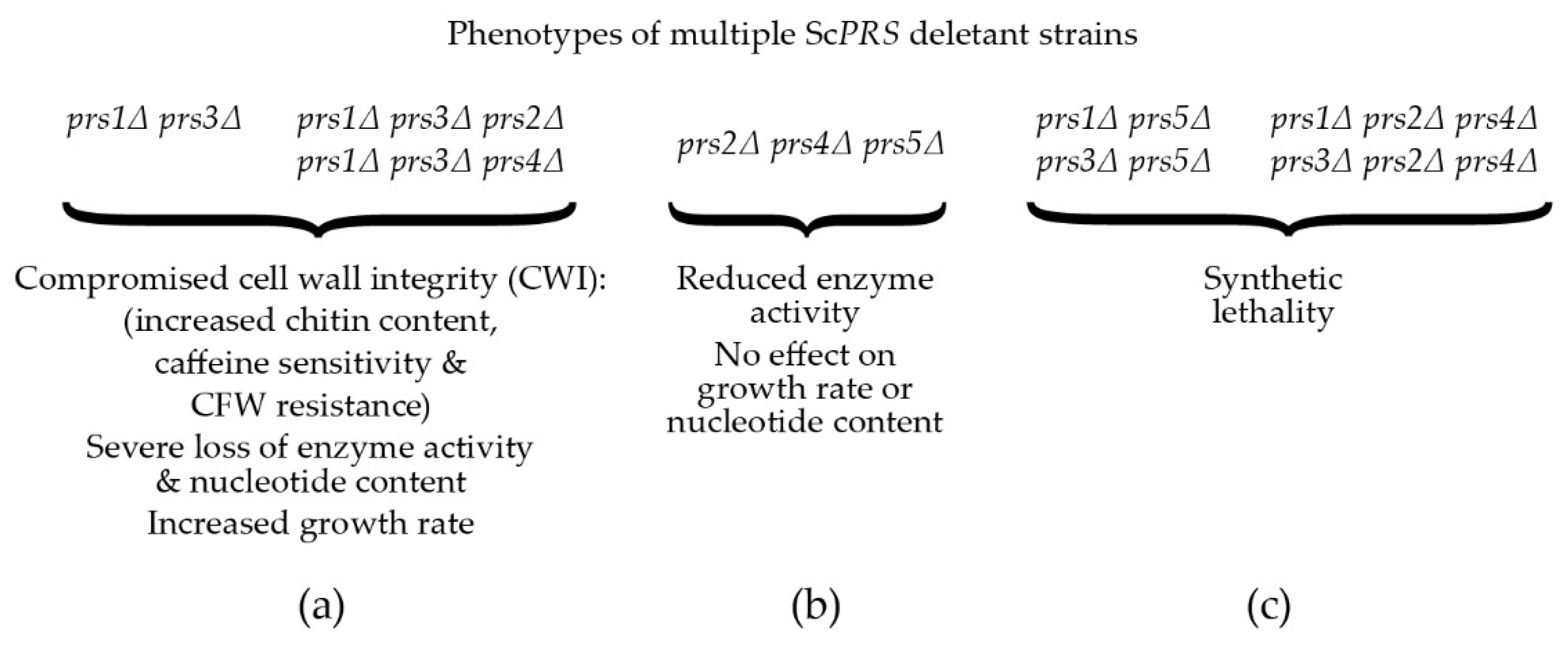
Figure 3. Compromised phenotypes, PRPP synthetase activity and nucleotide content encountered in ScPRS multiple deletant strains: (a) phenotypes of defined ScPRS deletant strains. CFW = calcofluor white; (b) phenotypes of the triple deletant prs2Δ prs4Δ prs5Δ; (c) synthetic lethality encountered when either ScPRS1 or ScPRS3 is deleted from a prs5Δ strain or simultaneous deletion of ScPRS2 and ScPRS4 in combination with loss of either ScPRS1 or ScPRS3.
By performing a variety of diagnostic tests, including environmental exposure, e.g., heat shock, caffeine sensitivity, CFW (calcofluor white), Congo red and exposure to mating pheromone, researchers discovered that strains deleted for ScPRS1 or ScPRS3 are compromised in the maintenance of CWI [64][78][81][82]. The general architecture of the MAPK (mitogen-activating protein kinase) pathway in eukaryotes is illustrated in Figure 4a. Excellent reviews of the yeast CWI pathway and the extent of its influence on yeast physiology have been published [83][84][85][86][87][88][89][90][91]. ScPRS mutant strains have consistently shown varying degrees of sensitivity to the purine analogue, caffeine (1,3,7-tri-methylxanthine), thus compromising CWI signalling via the MAPK (mitogen-activated protein kinase) [64][74][79][82][92]. It should be noted that caffeine not only interferes with the integrity of the cell wall, but also possibly by the inhibition of mTOR (mechanistic Target Of Rapamycin) mTORC1 and mTORC2, well-known regulators of eukaryotic cellular metabolism [93][94][95].
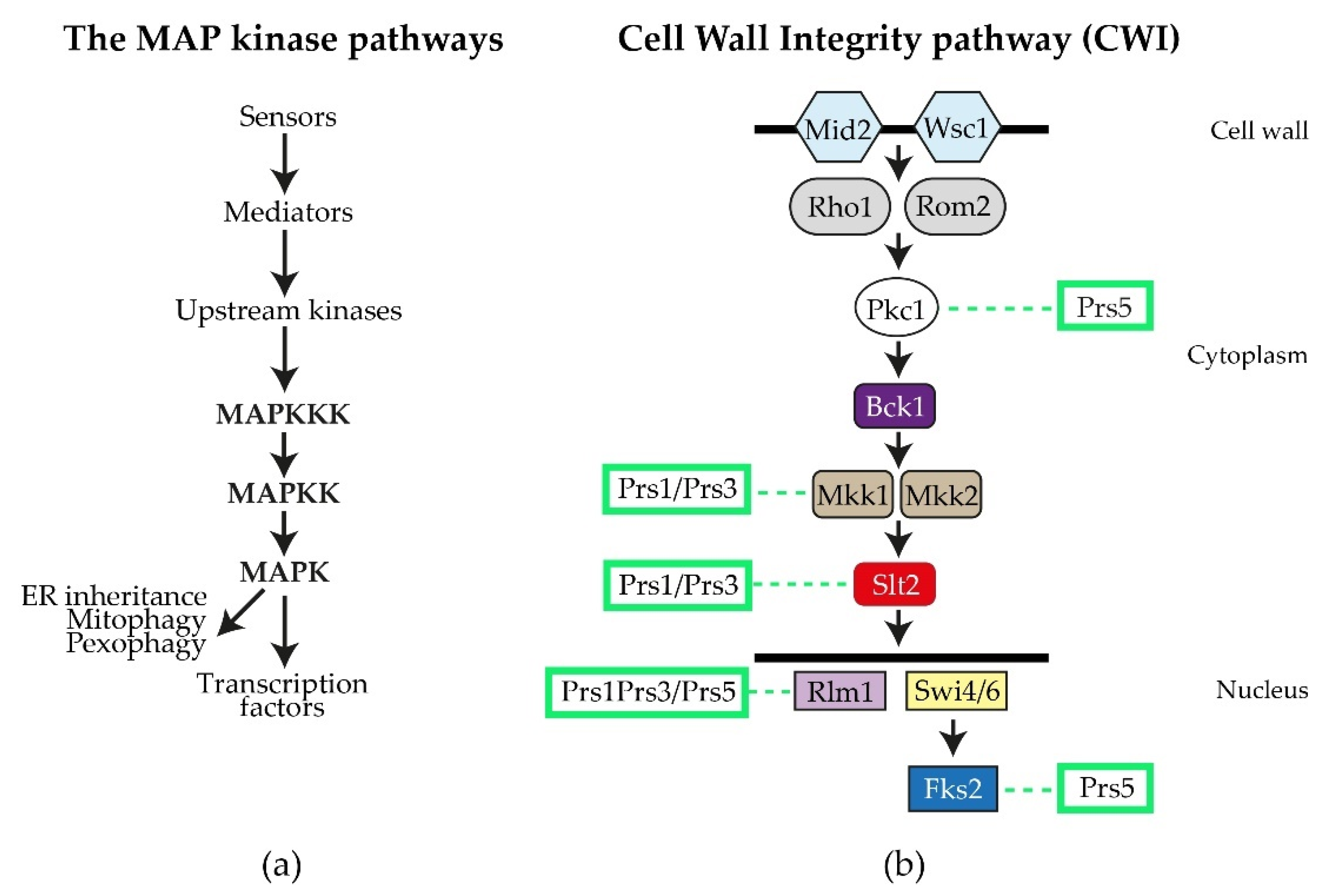
Figure 4. The S. cerevisiae CWI (cell wall integrity) pathway: (a) the general flow of information for MAP kinase pathways; (b) schematic diagram of the yeast CWI pathway. The two sensory proteins Mid2 and Wisc1 span the cell wall and are responsible for receiving signals, e.g., heat shock, that are transmitted to the upstream kinase Pkc1 by the mediators Rho1 and Rom2. Pkc1 then activates Bck1 (MAPKKK), which in turn activates Mkk1/Mkk2 (MAPKK), permitting the activation of Slt2 (MAPK). Slt2, in its phosphorylated state, can enter the nucleus, where it influences gene expression by targeting transcription factors, such as the MADS-box transcription factor Rlm1 and the SBF (Swi4/Swi6) transcriptional complex, as well as impacting on polarized growth and processes associated with mitophagy, pexophagy and ER inheritance. Fks2 is an alternative catalytic subunit of the β-1,3-D-glucan synthase whose expression is regulated by cell wall stress and Rho1 [83][84]. The interactions of Prs polypeptides with components of the CWI pathway or their impact thereon, are collated from the following references [64][74][78][79][80][84][92][95][96][97][98][99] and indicated by dashed lines.
Following gene duplication, yeast can sacrifice sources of PRS enzyme activity caused by the insertions, e.g., NHR1-1 and NHR5-2, since survival depends on the association of two Prs polypeptides, one containing at least one NHR and the other without an NHR. A concrete example of this is the Prs1/Prs3 heterodimer, which is the most important of the three minimal functional units required for yeast viability [27][75]. The insertion NHR1-1 in Prs1 disturbs the catalytic flexible loop, thus increasing its distance from the ribose-5-phosphate loop (Figure 2) and impacting its enzyme properties negatively but still permitting an interaction of Prs1 with the components of the CWI pathway, viz., the MAP kinase, Slt2/Mpk1 [92], a functional homologue of human ERK5 (extracellular signal-regulated kinase) [100][101], and Mkk1 (mitogen-activated protein protein-kinase) [78][81][102]. A comparable situation exists for Prs5, which, in combination with either Prs2 or Prs4, provides the two remaining minimal functional units essential for cell viability. The three insertions referred to above, explain the necessity of retaining at least one of the three minimal functional units, Prs1/Prs3, Prs2/Prs5 or Prs4/Prs5, since only then can the production of PRPP and the maintenance of CWI in the yeast cell be guaranteed. Furthermore, it also explains why simultaneous deletion of ScPRS1 and ScPRS5 or ScPRS3 and ScPRS5 is synthetically lethal since neither PRPP production nor maintenance of CWI is sustained (Figure 3c).
By Western blotting with human anti-phospho MAPK antibodies, which recognize S. cerevisiae phosphorylated Slt2, researchers demonstrated that strains lacking ScPRS2, ScPRS4 or ScPRS5 can activate Slt2 in response to heat stress in a pattern similar to that of the WT. However, the deletion of ScPRS1 or ScPRS3 causes constitutively phosphorylated Slt2 in the absence of heat stress. In a prs1Δ strain, there was a strong signal at time zero, which, after heat shock, was maintained over a period of four hours before falling to zero. A prs3Δ strain can maintain and increase the level of phosphorylated Slt2 upon exposure to heat stress [78][80][81]. The difference in the pattern of heat-induced Slt2 phosphorylation response in a prs3Δ strain is, in fact, the response of a prs1Δ prs3Δ, since it has been demonstrated that deletion of ScPRS3 or only its 284KKCPK288 motif, which researchers designated NHR3-1 (Figure 2), results in simultaneous loss of Prs1 [92]. The sudden disappearance of the phosphorylated form of Slt2 seen in the ScPRS1 at five hours of heat exposure suggested an inability of this strain to endure stress over a longer period, possibly due to the breakdown of the Prs1/Prs3 heterodimer [80][81].
Researchers have shown by co-immunoprecipitation that NHR1-1 interacts with the MAP kinase, Slt2, of the CWI pathway only when Slt2 is phosphorylated, thus providing more evidence for linking PRPP synthesis to the maintenance of CWI in yeast. A strain expressing non-phosphorylatable Slt2 (slt2(T190A, Y192F)) does not interact with Prs1; however, the kinase-dead Slt2 mutant (slt2(K54R)) still interacts with Prs1 [79][103]. It has also been possible to demonstrate that Prs1 is bifunctional since a measurable increase in the strength of the Y2H interaction between prs1(ΔNHR1-1) and Prs3 results in a 50% increase in PRS activity in comparison to the WT. A lower, albeit possibly more accurate factor, 15%, is measured when comparing PRS activity with strains carrying plasmid-borne versions of WT Prs1 and prs1(ΔNHR1-1). The inability of a prs1(ΔNHR1-1) strain to increase Rlm1 expression in response to heat stress while maintaining enzyme activity indicates, for the first time, that the two metabolic functions—provision of PRPP and maintenance of CWI—are separable and the presence of NHR1-1 is not essential for the formation of the Prs1/Prs3 heterodimer, as demonstrated by a stronger Y2H interaction with Prs3 [92].
2.5. Yeast Genocopies of PRPS-Associated Human Neuropathies Interfere with CWI
Prs2, Prs3 and Prs4 of yeast and hPRPS1 proteins share a sequence similarity of approximately 70%, which drops to 53% in Prs1 on account of the presence of NHR1-1 (Clustal Omega). Genocopies of mutations in ScPRS1 associated with human neuropathies were created to examine their impact on yeast physiology. Missense mutations in the hPRPS1 gene can lead to a gain-of-function associated with PRPS1 superactivity [104][105][106][107] or a loss-of-function [51][104]. Arts syndrome [47][48] and Charcot-Marie-Tooth disease-X5 (CMTX5) belong to the latter category [51]. Genocopies in ScPRS1 of CMTX5 (c.343C > A + c.344T > C (p.L115T) (cf. p.M115T (c.344T > C) in hPRPS1) and Arts syndrome (c.398A > C (p.Q133P) (cf. p.Q133P (c.398A > C) in hPRPS1) were created. These genocopies are located at 86 and 68 aa, respectively upstream of NHR1-1 in ScPRS1. Furthermore, mutations of the divalent-cation binding site alone or in combination with the ribose-5-phosphate binding site were created [74]. All constructs tested retain NHR1-1 and can increase Rlm1 expression at 37 °C, albeit to varying degrees (Figure 5a). Rlm1 expression in the presence of the genocopies p.L115T and p.Q133P was increased with respect to the WT at ambient temperature, indicating that these mutations may influence the folding of Prs1, which does not interfere with NHR1-1/Slt2 interaction [74]. A similar response of Rlm1 expression was observed when researchers tested the effect of mutating the divalent cation-binding site at position H130A (c.388C > G + c.389A > C) and the ribose-5-phosphate binding site, D326A (c.977G > C), singly and in combination, namely, an increased Rlm1 response at 30 °C, which increased further following incubation at 37 °C, suggesting that the folding of the mutated Prs1 polypeptides is altered in such a way that NHR1-1 can interact more readily with Slt2, possibly on account of their locations external to each monomer of the hexameric complex of B. subtilis PRPP synthetase complex [21]. Both NHR1-1 and p.H130A are essential for overcoming the synthetic lethality of a prs1Δ prs5Δ strain, whereas Prs1 with an altered ribose-5-phosphate binding site, p.D326A, can rescue the synthetic lethality [74][103]. The mutations p.H130A and p.Q133P are separated by only two aa but have significantly different levels of Rlm1 expression at 30 °C, suggesting that the helix breaker mutation, p.Q133P, has a more profound effect on protein folding. The Rlm1 expression data follow the same pattern as PRS activity (Figure 5b) [74]. Strains carrying these genocopies could suppress the synthetic lethality of the double mutant prs1Δ prs5Δ at 30 °C but did not restore growth at 37 °C. However, the inclusion of 1 M sorbitol as an osmotic stabilizer permitted growth at 37 °C, except for the strain carrying the genocopy of the mutation p.Q133P associated with Arts syndrome. Examining these rescued strains for their caffeine sensitivity indicated that in the absence of an osmotic stabilizer, growth was possible at 30 °C in 2 mM caffeine, although again, the genocopy p.Q133P (Arts syndrome) grew less well than the others. When 1 M sorbitol was included in the media, growth was restored at 30 °C at a concentration of 3 mM caffeine [103].
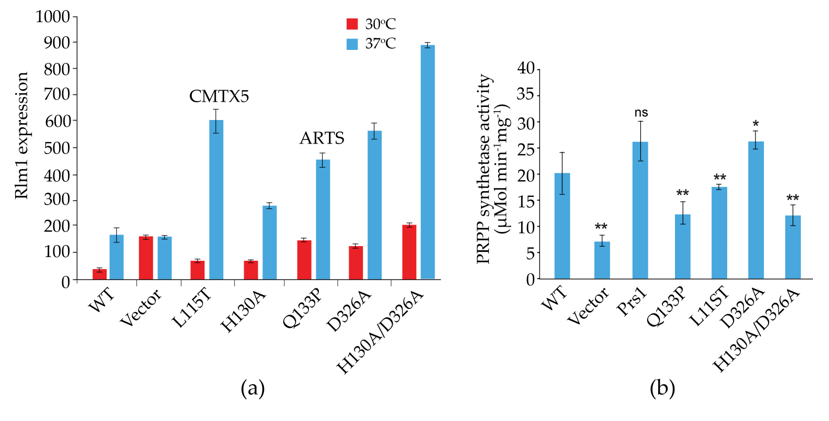
Figure 5. Influence of genocopies p.L115T and p.Q133P in ScPrs1, which correspond to the human neuropathies CMTX5 (p.M115T) and Arts syndrome (p.Q133P), respectively, and characteristic motifs of Prs1 either singly or in combination: H130A, divalent cation-binding site; D326A, PRPP-binding site/Ribose-5-phosphate binding site. It should be noted that the locations of the mutations H130A and D326A differ from those in Figure 2, since the length of Prs1 (427 aa) is increased with respect to Prs2 (318 aa) on account of the insertion of NHR1-1; (a) on Rlm1 expression following incubation at 30 °C and 37 °C, all values are significantly different from the WT at p < 0.001 (n = 12-20); (b) on PRS activity. p-values: * = p ≤ 0.05; ** = p ≤ 0.05, ns = not significant.
3. Human PRPS and Associated Disorders
In humans, the three isoforms of PRS, hPRPS1 (HGNC 9462), hPRPS2 (HGNC 9465) and hPRPS3 (HGNC 9463) [15][44][108][109][110][111][112], share high aa sequence similarity, namely 98.7% between hPRPS1 and hPRPS2, 97.2% between hPRPS1 and hPRPS3 and 96.5% between hPRPS2 and hPRPS3 (EMBOSS Needle). Human PRPS1 exists in two isoforms. The PRPS1 transcript variant 1 mRNA, RefSeq NM_002764 contains seven exons and gives rise to a protein RefSeq NP_002755 with a MW of 34.8 kDa. The hPRPS1 transcript variant 2 mRNA, RefSeq NM_001204402 gives rise to a shorter protein variant identical to the last 114 aa of PRPS1 isoform 1 (RefSeq NP_001191331) [112]. For hPRPS2, there are also two transcript variants 1 and 2 mRNAs (NM_001039091 and NM_002765), which differ only in length by nine nucleotides due to alternative splicing giving rise to two proteins, one of 321 aa and the other of 318 aa (RefSeq NP_001034180 and NP_002756) [43][45].
There are three classes of PRS enzymes, viz. I, II and III, with the hPRPS1 belonging to class I, which require inorganic phosphate (Pi), SO42− and Mg2+ for enzymatic activity, are dependent on ATP or dATP as the diphosphoryl donor and are allosterically regulated by ADP, AMP and GDP [3][21][113]. Class II enzymes are specific to plants, and they accept a wide range of diphosphoryl donors (ATP, dATP, CTP, GTP, and UTP) and are not allosterically inhibited by ADP or dependent on Pi for their activity [114][115]. Class III enzymes are a novel class of PRPP synthetase found in the archaeon Methanocaldococcus jannaschii, which utilize ATP and dATP as a diphosphoryl donor and whose activation is solely dependent on Pi. They are inhibited by ADP in a non-allosteric manner but appear to be competitively inhibited by ATP [116]. The PRPP synthetase from the thermophilic archaeon Thermoplasma volcanicum is an interesting case since on the basis of structure, it has a close similarity to M. jannaschii and therefore, has been designated as a class III PRPP synthetase, in spite of lacking an allosteric site and existing as a dimer [117]. In addition to the biochemical characterization of PRPP synthetases, they can also be classified according to their three-dimensional structures: class I enzymes have a hexameric quaternary structure found in mammals and bacteria; class II enzymes, including PRPP synthetase isozyme 4 from spinach, have a homo-trimer quaternary structure; and the class III enzyme from M. jannaschii is tetrameric [116]. The crystal structure of B. subtilis PRS (PDB: 1DKR) [21] has been known for twenty years and has provided the basis for structural analysis of hPRPS1 (PDB: 2H06) and is a homo-hexamer consisting of three tightly interacting homodimers [113]. Each of the homodimers contains a catalytic site and two allosteric sites, I and II, whereas, in the B. subtilis PRS, there is only one allosteric site [21][113]. The reactive site binds both ATP and ribose-5-phosphate and is located at the interface of two domains within one homodimer. The allosteric site I for ADP and Pi is positioned at the interface of the three homodimers of the hexameric structure, whereas the second, novel allosteric site II occupies a position at the interface of two monomers within one homodimer. The crystal structure of hPRPS1 reveals a complex with SO42− at the allosteric site II, thus positively impacting on the stability of the catalytic flexible loop and preventing the entry of the inhibitor ADP while maintaining an open conformation for the entry of ATP and activation of the enzyme [113][118].
Human PRPS1 and PRPS2 are class 1 PRPS enzymes that, despite their high degree of sequence similarity, differ in function and regulation. It has been observed that PRPS1 is expressed constitutively, whereas PRPS2 is expressed inter alia in colorectal cancer (CRC) metastasis and hepatocellular carcinoma (HCC) [119][120]. In addition to hPRPS1 and hPRPS2, human PRPS-associated proteins (PAPs) (PAP39 (hPRPSAP1, HGNC 9466) at position 17.q25.1 and PAP41 (hPRPSAP2, HGNC 9467) at position 17p11.2 have been discovered [13][17]. Human PAPs share high amino acid sequence similarity and interact physically with hPRPS1 [121][122][123][124][125]. However, PAPs, PRSPSAP1 and PRPSAP2 do not contain an ATP-catalytic binding site and, therefore, cannot be directly involved in the phosphoribosyl transferase reaction but may act as negative regulators of PRPP synthetase [13][17]. It should be noted that the insertions in rat PRSPSAP1 and rat PRPSAP2 are at similar positions to NHR1-1 of ScPRS1 and NHR5-2 of ScPRS5 (cf. Figure 2) [125]. Two processed pseudogenes for hPRPS1 (HGNC 39427 and HGNC 9464) are located on chromosomes 2q24.3 and 9q33.3, respectively, and both are transcribed but not translated. Human PRPS1-related diseases can be characterized by a continuous spectrum of features spanning neuropathies, hearing loss, optic atrophy, ataxia, cognitive impairment, and recurrent upper-respiratory infections [104]. It is possible to correlate mutations in hPRPS1 with the severity of the disease, which ranges across varying degrees of loss of PRPS activity attributable to minor changes in the architecture of the hexameric structure. For instance, PRPS activity in the neuropathy CMTX5 patients is reduced in comparison to patients presenting with DFN2-associated hearing loss because of the nature of the local structural changes of the enzyme. There is also the possibility that epigenetic modification affects hPRPS1 expression [126][127]. The neurological symptoms of hPRPS-related diseases may be associated with the degree of myelination of neuronal structures since the synthesis of myelin is dependent on lipid esters of pyrimidine nucleotides and CDP-choline and requires SAM (S-adenosylmethionine) [104]. Researchers have observed that in the prs1Δ strain, the combined CDP and CTP levels were extremely low [26][76][80], only 12% of the level in the WT, thus connecting PRPP synthesis to both de novo and salvage synthesis of phospholipids which require CTP. The osmotic sensitive phenotype of a prs1Δ strain is rescued by overexpression of CTP synthetase [80][128]. Neurons have a high energy demand and are therefore susceptible to any reduction in GTP and ATP supply. There is a parallel scenario in yeast that CTP pools are important for maintaining the supply of CDP-choline and CDP-ethanolamine for phospholipid biosynthesis, essential for membrane integrity [104][129]—a further argument for the use of yeast as a eukaryotic model organism. The role of pyridine nucleotides in energy production cannot be ignored since they are involved in energy storage, cell signalling, nucleic acid structure and enzyme cofactors, e.g., NAD+ [104]. The conflicting evidence that PRPS1 superactivity can lead to purine depletion and overproduction of uric acid reflects the spectrum of mutations encountered in the product of the hPRPS1 gene, which can affect local enzyme structure or impinge on the integrity of the quaternary structure and the stability and overall activity of the enzyme.
3.1. PRPP Synthetase Superactivity: hPRPS1 (OMIM 300661)
Missense mutations of hPRPS1, the more highly expressed isoform of the two hPRPS genes, lead to gain-of-function mutants [46][104][130]. These changes cause critical alterations in PRPS levels that may have a significant influence on many life processes, such as nucleotide biosynthesis and cell signalling. The gain-of-function of hPRPS1 results in superactivity of the enzyme associated with hyperuricaemia and hyperurocosuria—overproduction and accumulation of uric acid in the blood and urine, respectively. Overproduction of uric acid can lead to gouty arthritis in joints and impairment of kidney function because of excessive production of purine nucleotides and their subsequent conversion to uric acid [50] and references therein [105][107][131][132]. Specifically, PRPS1 superactivity is a rare X-linked disorder of purine metabolism that results in an increase in enzyme activity due to regulatory defects or alteration in catalytic properties [14][130][133][134][135][136][137]. Overexpression of hPRPS1 with altered kinetic enzyme characteristics has been observed in white cells, erythrocytes and fibroblasts of patients [131][136][137] and is characterized by several physiological disorders, including, in addition to gouty arthritis, hearing impairment or deafness, hyperuricaemia, hyperuricosuria, urolithiasis in men, ataxia, hypotonia, developmental delay, intellectual impairment and recurrence of infectious diseases in the upper respiratory tract [104][105][131][138][139][140][141]. Since the hPRPS1 gene is located on the X-chromosome, if one of the mother’s two X-chromosomes carries the mutation for PRPS superactivity and this chromosome is inherited by the son, he will suffer this disease, and he can pass the affected chromosome to a daughter. The daughter will not necessarily display symptoms since she may have inherited an unaffected chromosome from her mother. Women are less likely to suffer from PRPS1 superactivity since random X-chromosome inactivation [142][143] may reduce the dosage or eliminate the mutated version of the hPRPS1 gene.
Patients with severe or extremely high PRPS1 superactivity may exhibit one or more of the following nine missense PRPS1 mutations, p.D52H (c.154G > C), p.L129I (c.385C > A), p.A190V (c.569C > T), p.H193Q (c.579C > G) [133], p.N114S (c.341A > G) and p.D183H (c.547G > C) [104][133], p.H193L (c.578A > T) [132][133], p.G174V (c.521G > T) [144] and p. G174R (c.520G > A) [145]. Specifically, the missense mutations of p.D52H and p.L129I result in the disruption of local structure close to the allosteric sites and therefore inhibit feedback regulation [104][133][146]. The p.D52H mutation causes destabilization of the enzyme structure around Asp52 and directly disturbs allosteric site I [104][106][127], thus reducing the sensitivity to the inhibitor ATP in vivo [146], whereas the p.L129I mutation leads to steric hindrance with both the protein backbone at Ala131 and Ile134 disturbing allosteric site II [104], leading to a neuropathy [127][133].
The other missense mutations are located in the homodimer interface, thereby disrupting the homodimer and affecting the allosteric sites [105][132]. The p.N114S mutation also destabilizes the hydrogen bonding of the homodimer. The p.A190V mutation leads to disruption of the hydrophobic environment in the homodimer interface, whereas p.D183H and p.H193L/Q mutations break the hydrogen bond interactions between Asp183 of one homodimer partner and His193 of the other polypeptide chain of the homodimer, thereby destabilizing the homodimer interface [104][127]. Interestingly, the hPRPS1 mutations p.D183H and p.N114S in both hemizygous males and affected females caused neurological deficiency, e.g., sensorineural deafness [50][106], whereas p.D52H leads to increased production of PRPP levels and induces hyperuricaemia and gout [146]. In males, PRPS1 superactivity resulted in an alteration in the allosteric control of PRPP synthesis with high purine nucleotide and uric acid production [17]. However, the study of Zikánová et al. (2018) [145] showed that female carriers of PRPS1 superactivity had elevated levels of uric acid and gout since random X-chromosome inactivation may result in them retaining only the mutated allele p.G174R (c.520G > C).
3.2. Reduced PRPP Synthetase in Humans
Reduced activity in PRPS1 is associated with neurological disorders, such as Arts syndrome, Charcot-Marie-Tooth disease type 5 (CMTX5), X-linked syndromic/nonsyndromic sensorineural deafness (DFN2/DFNX2) and retinal dystrophy.
3.2.1. Arts Syndrome
This disorder (OMIM 301835) is associated with X-linked missense mutations in hPRPS1, resulting in loss-of-function of PRPS1. It affects mainly males with a prevalence of less than 1 in 106 and is lethal, with symptoms appearing before two years of age. Unfortunately, 80% of patients die in childhood. It is a severe phenotype caused by the missense mutation in c.455T > C (p.L152P), which was first identified in a Dutch family. A further missense mutation c.398A > C (p.Q133P) was identified in an Australian family [47][48]. MD revealed that the p.Q113P variant destabilizes the allosteric site II and the ATP binding pocket of hPRPS1 and that p.L152P alters only the ATP binding site [48]. An autopsy of the Dutch Arts syndrome patient revealed a total lack of myelin in the posterior columns of the spinal cord, and a sural nerve biopsy of the Australian patient showed mild paranodal demyelination. Arts syndrome is characterized by profound delayed motor development, early-onset hypotonia, hearing impairment, mental retardation, optic atrophy, intellectual disability, a compromised immune system, recurrent infection, and early childhood mortality [47][48][104].
Maruyama et al. (2016) [147] also reported a transient neurogenic proximal weakness and loss of muscle strength caused by the mutation p.H123D (c.367C > G) in Arts syndrome patients. The premature death of Arts syndrome patients is most commonly due to infections of the upper respiratory tract [47]. In this disorder, low serum uric acid concentrations and undetectable hypoxanthine with normal range xanthine and uric acid concentrations in the urine purine profile have been observed [48]. In affected males, PRPS enzyme activity is significantly decreased, while obligate female carriers show mild symptoms [148]. PRPP synthetase activity, as measured in fibroblasts from the affected members of the Dutch family, was reduced 10-fold in comparison to the activity from controls [47]. In a recent study, Puusepp et al. (2020) [149] reported no hearing loss and normal levels of both serum uric acid, purines and pyrimidines in a hemizygous variant c.130A > G (p.I44V) of the hPRPS1 gene in a 6-year-old male patient presenting with clinical features of Arts syndrome, as described above. However, a marked reduction in PRPS1 activity in erythrocytes was recorded in this patient, emphasizing the necessity of testing for all parameters associated with Arts syndrome. It is noteworthy that the p.I44 residue is conserved in mammals and amphibia, Drosophila spp., Caenorhabditis elegans and S. cerevisiae.
Another mutation, c.424G > C (p.V142L), was found in a patient presenting with classical symptoms, including hyperuricaemia and hyperuricosuria but with no gouty arthritis, developmental delay, hypotonia, and bilateral hearing loss, indicative of both the severe form of hPRPS1 superactivity and symptoms of Arts syndrome, e.g., recurrent respiratory infections, myopia, glaucoma and motor neuropathy [139]. This valine-to-leucine change arising from a transversion is predicted to alter the allosteric sites I and II and the ATP binding site, thus impairing hPRPS1 inhibition and resulting in hPRPS1 overexpression. Furthermore, this substitution is postulated to influence the interaction of Val142 with Lys100 in a flexible loop, which is part of the ATP-binding site of the other PRPS1 subunit of the homodimer. The two-fold effect of this mutation on nucleotide concentrations is reflected in the difference in PRPS activity measured in fibroblasts and postmitotic cells, e.g., erythrocytes and brain cells. In the former, there is increased PRPS1 activity due to loss of feedback inhibition, whereas in the latter, there is loss of PRPS1 activity over time. This combination of gain-of-function and loss-of-function of PRPS1 activity in the same patient was revealed by molecular modelling and enzyme activity measurement in different cell types and illustrated the range of features associated with an imbalance of PRPP synthetase underlying the clinical features of various non-syndromic postlingual hearing loss neuropathies.
In summary, there are several manifestations, e.g., PRPS1 superactivity and recurrent infections, which can be traced back to the p.V142L mutation resulting in intermediate phenotypes lacking the severity of those associated with Arts syndrome (p.Q133P or p.L152P). There was a difference in PRPP synthetase activity as measured in erythrocytes and fibroblasts in the proband p.V142L, which could be due to the altered regulatory properties of the enzyme. Supporting evidence for this suggestion is that the GDP/GTP levels in the proband’s erythrocytes were reduced in comparison to the controls, but ATP/ADP levels were normal. Unfortunately, there was no data concerning GTP/GDP levels in fibroblasts, although enzyme activity in fibroblasts carrying the mutation did respond to the level of phosphate, whereas the control did not. It could be that intracellular purine levels are sensed by an as-yet-unknown mechanism(s) involving GTP interaction with mTORC1 via Rheb [150].
Overlapping phenotypes are also described in a male/female sibling study [127]. A male subject, aged 36, p.Q277P (c.830A > C), displayed overlapping features of CMTX5 and Arts syndrome, prelingual hearing loss, recurrent severe infections and progressive visual loss due to optic atrophy, as well as undetectable PRPS1 activity in erythrocytes. His sister had reduced PRPS1 activity in addition to prelingual nonsyndromic hearing loss and deafness (DFN2/DFNX1), whereas their mother had normal PRPS1 activity and no hearing loss. Brain MRI (magenetic resonance imaging) of the two siblings revealed cerebellar atrophy. X-chromosome inactivation was extremely skewed in the sister but only moderately skewed in the mother. Recently, in 2021 [151], a novel Arts syndrome hPRPS1 deficiency missense mutant p.R84T (c.250C > T) was reported, which disturbed the interface regions between the subunits, thus negatively impacting the ligand binding of the allosteric site I. An Australian male patient presented in his first year with symptoms congruent with Arts syndrome, including congenital sensorineural deafness, recurrent severe infections, visual impairment inter alia with substantially reduced hPRPS1 activity in erythrocytes. It was decided with parental consent to start feeding studies with SAM (S-adenosylmethionine) and NR (nicotinamide riboside) at the age of three. Unlike most purines, SAM can cross both the gut and the blood-brain barrier and has been used successfully in the treatment of depression and dementia [152]. It was also shown that SAM, which can supply ATP and GTP independently of PRPP [104], may alleviate some of the symptoms of patients with Arts syndrome. NR can form NADP(H) independently of PRPP by virtue of NR kinases 1 and 2 to produce NMN (nicotinamide mononucleotide), which is adenylated to NAD+ [153][154]. Shortly after commencing treatment, the intervals between respiratory infections increased and recovery was improved. The patient’s muscular strength, speech, ability to play and overall well-being improved markedly. No adverse treatment effects were recorded, and homocysteine/methionine concentrations were normal. Despite these improvements, audio-visual impairment and ataxia persisted, but nevertheless, the treatment reduced the strain on the patient and his family. ATP and GTP improved sustainably, but NAD+ measurements could not be repeated with higher levels of NR [104][151].
4. Roles of PRPS in Cancer
A recent development is the discovery of the involvement of PRPP synthetases 1 and 2—hPRPS1 & hPRPS2—in cancer progression, management, and treatment. Human PRPS1 and PRPS2 have been identified in acute lymphoblastic leukaemia (ALL) as the major cause of drug resistance to the prodrugs thioguanine and thiopurine, which are used in cancer therapy [155][156][157][158]. The way in which cancer cells become resistant to the prodrugs is through the acquisition of hyper-activating hPRPS1 mutations, e.g., c.568G > A (p.A190T), p.A190V, c.340A > G (N114D), p.D183H, c.521G > A (p.G174E), c.528A > T/C (p.K176N), c.571C > T + c.573G > T/C (p.L191F), c.308G > C (p.S103T) ([159] and other mutations cited therein). All these mutations destabilize the dimer interface, causing the loss of ADP and GDP allosteric inhibition. Large-scale sequencing of DNA from patients with therapy-resistant mutations in relapsed ALL showed the presence of hPRPS2 hyper-activating mutations c.400G > A (p.A134T) and c.523G > A (p.A175T) [156]. hPRPS1 and hPRPS2 have been implicated in the metastasis and proliferation of cancer [159][160]. Mammalian PRPS2 is associated with oncogene Myc-driven tumorigenesis activation [52] and promotes metastasis of neuroblastoma cancer [160]. The translationally regulated enzyme PRPS2 may play a role in the proliferation of oncogenic signalling-driven cancers. This is due to an eIF-4E cis-regulatory pyrimidine-rich translational element (PRTE) found in mPRPS2 and hPRPS2 [52][161], but not in the corresponding PRPS1 genes, which renders PRSP2 resistant to feedback inhibition by ADP and GDP and may also explain the therapy-resistant human cell lines associated with ALL [156]. The distinguishing features of the PRPS2 isoform are found in many translationally regulated transcripts because of mTOR hyperactivation, allowing nucleotide and protein biosynthesis to increase concomitantly with higher protein synthetic capacity protein of cancer cells [52][162][163]. PRPS2-knockout mice are viable and fertile with no discernible developmental defects, which supports the hypothesis that mPRPS2 is not essential for development but contributes to Myc-dependent cancer formation. However, a recent publication [164] has correlated the depletion of murine PRPS2 by sh RNA lentivirus with hypospermatogenesis, suggesting that PRPS2 may be a potential biomarker for male infertility. Human PRPS2 may well be an attractive diagnostic target for Myc-driven cancers in osteosarcoma [165]. The c-Myc-PRPS2 interaction promotes metabolic reprogramming of biosynthetic processes leading to the proliferation of cancer cells. There is statistical evidence linking a longer survival time with negative PRPS2/p-mTOR staining in comparison to positive PRPS2/p-mTOR staining, implying that Myc-driven PRPS2 expression via the eukaryotic translation initiation factor 4E (eIF-4E) allowing interaction with mTOR results in tumour progression.
Overexpression of hPRPS1 elevates cancer cell proliferation and inhibits apoptosis in B-cell acute lymphoblastic leukaemia cell lines (B-ALL) as well as increasing the expression of Bcl-2, a gene in the mitochondrial-associated apoptosis pathway, at the RNA and protein levels [166]. The same authors suggested that hPRPS1 silencing has no effect on the regulators of the cell cycle but elevates apoptosis in B-ALL cell lines. He et al. (2017) [167] found that hPRPS1 silencing increases apoptosis in breast cancer. The expression of hPRPS1 and hPRPS2 is reduced by hypoxia in glioma cells. Furthermore, the expression of PRPS1, PRPS2, PRPSAP1 (PAP39) and PRPSAP2 (PAP41) genes in U87 glioma cells is not inhibited by ERN1 (EC 2.7.11, endoribonuclease activity of endoplasmic reticulum to nuclei-1). Endoplasmic reticulum-induced stress via tunicamycin in glioma cells, together with the suppression of ERN1, downregulates PRPS1 and PRPS2 gene expression levels [168]. The combined deletion of PRPS1 and PRPS2, PPAT (PRPP-amidotransferase, EC 2.4.2.14) (HGNC 9238) and CAD (carbamoylphosphate synthetase II, EC 6.3.5.5) (HGNC 1424) suppresses cell proliferation in Kaposi’s sarcoma-associated herpes virus (KSHV), by redirecting glutamine and asparagine, which provide the nitrogen source for purine and pyrimidine biosynthesis from the TCA cycle to the biosynthesis of nucleotides and non-essential amino acids, suggesting potential therapeutic application for this pathway [169].
Interestingly, in a recent study conducted by Miao and Wang (2019) [119], it has been reported that hPRPS2 promotes the movement and invasion of colorectal cancer cells (CRC), suggesting that the upregulation of PRPS2 observed in CRC was induced by the MYC proto-oncogene, thus implicating PRPS2 in CRC metastasis. Previously, Qiu et al. (2015) [54] demonstrated that the mRNA and protein levels of hPRPS1 are upregulated in CRC. Knockdown of hPRPS1 or upregulation of the micro-RNA, miR-124, a tumour suppressor, leads to reduced DNA synthesis and cell proliferation in CRC cell lines, revealing potential therapeutic targets. There are miRNA integrated signatures for hPRPS1 mRNA and hPRPS2 mRNA interactions, which may represent potential biomarkers and therapeutic targets for CRC [170].
MD simulation of four missense mutations p.D52H, p.M115T, p.L152P and c.607G > C (p.D203H) in hPRPS1 predicted to be disease-causing mutations [171], revealed that the CMTX5 mutation p.M115T affected protein stability and is consistent with previously published experimental results documenting 60% reduction in hPRPP synthetase activity due to the disruption of the ATP binding region and allosteric site I of the PRPS1 active site [51]. Interestingly, a further hPRPS1 missense mutation c.335T > C (p.V112A) spans the reference SNP (rs11541075) for the BOR (Branchio-Oto-Renal) syndrome. This syndrome, which is associated with hearing loss and other hearing abnormalities, in addition to abnormalities in kidney structure and function, may affect approximately 1 in 40,000 people (MedlinePlus). These data show the far-reaching effects of missense mutations in the hPRPS1 gene which, on reflection, are not unexpected in a gene whose product is required to produce essential metabolites involved in nucleotide synthesis as well as contributing to cellular signalling and energy metabolism [171][172].
KHK-A (ketohexokinase-A), a protein kinase that is a splicing variant of KHK-C, an enzyme involved in fructose metabolism, phosphorylates and activates hPRPS1 at T225 via the inhibition of ADP, AMP, and GDP by sterical blocking allosteric site I to ADP, thus leading to an increase in nucleotide synthesis and progression of HCC. The oncogenic transcription factor c-Myc upregulates hnRNPH1/2 expression, thereby favouring splicing from KHK-C to KHK-A which then phosphorylates PRPS1 at T225, increasing PRPP synthesis. By in vitro phosphorylation assays it was shown that KHK-A functions as a protein kinase rather than phosphorylating fructose, thus constitutively activating PRPP production in HCC which has been linked to a considerable risk of poor prognosis in HCC [173]. Clinical relevance has been demonstrated by immuno-histochemical comparative studies of matched tumour and non-tumour liver tissue samples supporting the molecular biological studies [173]. The upregulation of PRPS1 by KHK-A has also been shown to increase the growth of oesophageal squamous cell carcinoma [174]. Furthermore, the suppressed tumour formation of glioma CD133+ cells by reduction in PRPS1 expression is a result of increased cell apoptosis [175]. Screening of a miRNA-targeting database identified PRPS1 mRNA as being negatively regulated by the expression of miR-154 in glioma CD133+ cells [176]. This result has been echoed in neuroblastoma cells [177], adding weight to the potential of PRPS1 targeted therapy in glioblastoma multiforme and neuroblastoma cell proliferation.
This entry is adapted from the peer-reviewed paper 10.3390/cells11121909
References
- Kornberg, A.; Lieberman, I.; Simms, E.S. Enzymatic synthesis and properties of 5-phosphoribosylpyrophosphate. J. Biol. Chem. 1955, 215, 389–402.
- Khorana, H.G.; Fernandes, J.F.; Kornberg, A. Pyrophosphorylation of ribose 5-phosphate in the enzymatic synthesis of 5-phosphorylribose 1-pyrophosphate. J. Biol. Chem. 1958, 230, 941–948.
- Hove-Jensen, B.; Andersen, K.R.; Kilstrup, M.; Martinussen, J.; Switzer, R.L.; Willemoes, M. Phosphoribosyl Diphosphate (PRPP): Biosynthesis, Enzymology, Utilization, and Metabolic Significance. Microbiol. Mol. Biol. Rev. 2017, 81, e00040-16.
- Fox, I.H.; Kelley, W.N. Human phosphoribosylpyrophosphate synthetase. Kinetic mechanism and end product inhibition. J. Biol. Chem. 1972, 247, 2126–2131.
- Becker, M.A.; Ahmed, M. Cell type-specific differential expression of human PRPP synthetase (PRPS) genes. In Purine and Pyrimidine Metabolism in Man X. Advances in Experimental Medicine and Biology; Zoref-Shani, E., Sperling, O., Eds.; Springer: Boston, MA, USA, 2002; Volume 486, pp. 5–10.
- Hove-Jensen, B. Mutation in the phosphoribosylpyrophosphate synthetase gene (prs) that results in simultaneous requirements for purine and pyrimidine nucleosides, nicotinamide nucleotide, histidine, and tryptophan in Escherichia coli. J. Bacteriol. 1988, 170, 1148–1152.
- Jensen, K.; Dandanell, G.; Hove-Jensen, B.; Willemoës, M. Nucleotides, Nucleosides, and Nucleobases. EcoSal Plus 2008, 3, 1.
- Balasubramaniam, S.; Duley, J.A.; Christodoulou, J. Inborn errors of purine metabolism: Clinical update and therapies. J. Inherit. Metab. Dis. 2014, 37, 669–686.
- Balasubramaniam, S.; Duley, J.A.; Christodoulou, J. Inborn errors of pyrimidine metabolism: Clinical update and therapy. J. Inherit. Metab. Dis. 2014, 37, 687–698.
- Fasullo, M.; Endres, L. Nucleotide salvage deficiencies, DNA damage and neurodegeneration. Int. J. Mol. Sci. 2015, 16, 9431–9449.
- Villa, E.; Ali, E.S.; Sahu, U.; Ben-Sahra, I. Cancer cells tune the signaling pathways to empower de novo synthesis of nucleotides. Cancers 2019, 11, 688.
- Ullman, B.; Carter, D. Molecular and biochemical studies on the hypoxanthine-guanine phosphoribosyltransferases of the pathogenic haemoflagellates. Int. J. Parasitol. 1997, 27, 203–213.
- Tatibana, M.; Kita, K.; Taira, M.; Ishijima, S.; Sonoda, T.; Ishizuka, T.; Iizasa, T.; Ahmad, I. Mammalian phosphoribosyl-pyrophosphate synthetase. Adv. Enzym. Regul. 1995, 35, 229–249.
- Roessler, B.J.; Nosal, J.M.; Smith, P.R.; Heidler, S.A.; Palella, T.D.; Switzer, R.L.; Becker, M.A. Human X-linked phosphoribosylpyrophosphate synthetase superactivity is associated with distinct point mutations in the PRPS1 gene. J. Biol. Chem. 1993, 268, 26476–26481.
- Taira, M.; Iizasa, T.; Shimada, H.; Kudoh, J.; Shimizu, N.; Tatibana, M. A human testis-specific mRNA for phosphoribosylpyrophosphate synthetase that initiates from a non-AUG codon. J. Biol. Chem. 1990, 265, 16491–16497.
- Kita, K.; Otsuki, T.; Ishizuka, T.; Tatibana, M. Rat liver phosphoribosyl pyrophosphate synthetase: Existence of the purified enzyme as heterogeneous aggregates and identification of the catalytic subunit. J. Biochem. 1989, 105, 736–741.
- Becker, M.A. Phosphoribosylpyrophosphate synthetase and the regulation of phosphoribosylpyrophosphate production in human cells. Prog. Nucleic Acid Res. Mol. Biol. 2001, 69, 115–148.
- Krath, B.N.; Hove-Jensen, B. Organellar and cytosolic localization of four phosphoribosyl diphosphate synthase isozymes in spinach. Plant Physiol. 1999, 119, 497–506.
- Krath, B.N.; Eriksen, T.A.; Poulsen, T.S.; Hove-Jensen, B. Cloning and sequencing of cDNAs specifying a novel class of phosphoribosyl diphosphate synthase in Arabidopsis thaliana. Biochim. Biophys. Acta 1999, 1430, 403–408.
- Lin, X.; Kaul, S.; Rounsley, S.; Shea, T.P.; Benito, M.I.; Town, C.D.; Fujii, C.Y.; Mason, T.; Bowman, C.L.; Barnstead, M.; et al. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature 1999, 402, 761–768.
- Eriksen, T.A.; Kadziola, A.; Bentsen, A.K.; Harlow, K.W.; Larsen, S. Structural basis for the function of Bacillus subtilis phosphoribosyl-pyrophosphate synthetase. Nat. Struct. Biol. 2000, 7, 303–308.
- Baugh, L.; Gallagher, L.A.; Patrapuvich, R.; Clifton, M.C.; Gardberg, A.S.; Edwards, T.E.; Armour, B.; Begley, D.W.; Dieterich, S.H.; Dranow, D.M.; et al. Combining functional and structural genomics to sample the essential Burkholderia structome. PLoS ONE 2013, 8, e53851.
- Carter, A.T.; Narbad, A.; Pearson, B.M.; Beck, K.F.; Logghe, M.; Contreras, R.; Schweizer, M. Phosphoribosylpyrophosphate synthetase (PRS): A new gene family in Saccharomyces cerevisiae. Yeast 1994, 10, 1031–1044.
- Carter, A.T.; Beiche, F.; Narbad, A.; Hove-Jensen, B.; Schweizer, L.M.; Schweizer, M. Are all four yeast PRS genes essential? Biochem. Soc. Trans. 1995, 23, 621S.
- Carter, A.T.; Beiche, F.; Hove-Jensen, B.; Narbad, A.; Barker, P.J.; Schweizer, L.M.; Schweizer, M. PRS1 is a key member of the gene family encoding phosphoribosylpyrophosphate synthetase in Saccharomyces cerevisiae. Mol. Gen. Genet. 1997, 254, 148–156.
- Hernando, Y.; Parr, A.; Schweizer, M. PRS5, the fifth member of the phosphoribosyl pyrophosphate synthetase gene family in Saccharomyces cerevisiae, is essential for cell viability in the absence of either PRS1 or PRS3. J. Bacteriol. 1998, 180, 6404–6407.
- Wood, V.; Gwilliam, R.; Rajandream, M.A.; Lyne, M.; Lyne, R.; Stewart, A.; Sgouros, J.; Peat, N.; Hayles, J.; Baker, S.; et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002, 415, 871–880.
- DeSmidt, A.A.; Zou, B.; Grati, M.; Yan, D.; Mittal, R.; Yao, Q.; Richmond, M.T.; Denyer, S.; Liu, X.Z.; Lu, Z. Zebrafish Model for Nonsyndromic X-Linked Sensorineural Deafness, DFNX1. Anat. Rec. 2020, 303, 544–555.
- Pei, W.; Xu, L.; Varshney, G.K.; Carrington, B.; Bishop, K.; Jones, M.; Huang, S.C.; Idol, J.; Pretorius, P.R.; Beirl, A.; et al. Additive reductions in zebrafish PRPS1 activity result in a spectrum of deficiencies modeling several human PRPS1-associated diseases. Sci. Rep. 2016, 6, 29946.
- Hove-Jensen, B. Cloning and characterization of the prs gene encoding phosphoribosylpyrophosphate synthetase of Escherichia coli. Mol. Gen. Genet. 1985, 201, 269–276.
- Lau, N.S.; Makita, Y.; Kawashima, M.; Taylor, T.D.; Kondo, S.; Othman, A.S.; Shu-Chien, A.C.; Matsui, M. The rubber tree genome shows expansion of gene family associated with rubber biosynthesis. Sci. Rep. 2016, 6, 28594.
- Tang, C.R.; Yang, M.; Fang, Y.J.; Luo, Y.F.; Gao, S.H.; Xiao, X.H.; An, Z.W.; Zhou, B.H.; Zhang, B.; Tan, X.Y.; et al. The rubber tree genome reveals new insights into rubber production and species adaptation. Nat. Plants 2016, 2, 16073.
- Yu, H.; Zhang, Y.; Zhang, D.; Lu, Y.; He, H.; Zheng, F.; Wang, M. Identification of a Ribose-Phosphate Pyrophosphokinase that Can Interact In Vivo with the Anaphase Promoting Complex/Cyclosome. Int. J. Mol. Sci. 2017, 18, 617.
- Amalou, Z.; Bangratz, J.; Chrestin, H. Ethrel (Ethylene Releaser)-Induced Increases in the Adenylate Pool and Transtonoplast DeltapH within Hevea Latex Cells. Plant Physiol. 1992, 98, 1270–1276.
- Feng, Q.; Liu, Z.L.; Weber, S.A.; Li, S. Signature pathway expression of xylose utilization in the genetically engineered industrial yeast Saccharomyces cerevisiae. PLoS ONE 2018, 13, e0195633.
- Cunha, J.T.; Aguiar, T.Q.; Romani, A.; Oliveira, C.; Domingues, L. Contribution of PRS3, RPB4 and ZWF1 to the resistance of industrial Saccharomyces cerevisiae CCUG53310 and PE-2 strains to lignocellulosic hydrolysate-derived inhibitors. Bioresour. Technol. 2015, 191, 7–16.
- Cunha, J.T.; Costa, C.E.; Ferraz, L.; Romani, A.; Johansson, B.; Sa-Correia, I.; Domingues, L. HAA1 and PRS3 overexpression boosts yeast tolerance towards acetic acid improving xylose or glucose consumption: Unravelling the underlying mechanisms. Appl. Microbiol. Biotechnol. 2018, 102, 4589–4600.
- Cunha, J.T.; Soares, P.O.; Baptista, S.L.; Costa, C.E.; Domingues, L. Engineered Saccharomyces cerevisiae for lignocellulosic valorization: A review and perspectives on bioethanol production. Bioengineered 2020, 11, 883–903.
- Lucarelli, A.P.; Buroni, S.; Pasca, M.R.; Rizzi, M.; Cavagnino, A.; Valentini, G.; Riccardi, G.; Chiarelli, L.R. Mycobacterium tuberculosis phosphoribosylpyrophosphate synthetase: Biochemical features of a crucial enzyme for mycobacterial cell wall biosynthesis. PLoS ONE 2010, 5, e15494.
- Alderwick, L.J.; Lloyd, G.S.; Lloyd, A.J.; Lovering, A.L.; Eggeling, L.; Besra, G.S. Biochemical characterization of the Mycobacterium tuberculosis phosphoribosyl-1-pyrophosphate synthetase. Glycobiology 2011, 21, 410–425.
- Trefzer, C.; Rengifo-Gonzalez, M.; Hinner, M.J.; Schneider, P.; Makarov, V.; Cole, S.T.; Johnsson, K. Benzothiazinones: Prodrugs that covalently modify the decaprenylphosphoryl-D-ribose 2′-epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665.
- Donini, S.; Garavaglia, S.; Ferraris, D.M.; Miggiano, R.; Mori, S.; Shibayama, K.; Rizzi, M. Biochemical and structural investigations on phosphoribosylpyrophosphate synthetase from Mycobacterium smegmatis. PLoS ONE 2017, 12, e0175815.
- Taira, M.; Kudoh, J.; Minoshima, S.; Iizasa, T.; Shimada, H.; Shimizu, Y.; Tatibana, M.; Shimizu, N. Localization of human phosphoribosylpyrophosphate synthetase subunit I and II genes (PRPS1 and PRPS2) to different regions of the X chromosome and assignment of two PRPS1-related genes to autosomes. Somat. Cell Mol. Genet. 1989, 15, 29–37.
- Becker, M.A.; Heidler, S.A.; Bell, G.I.; Seino, S.; Le Beau, M.M.; Westbrook, C.A.; Neuman, W.; Shapiro, L.J.; Mohandas, T.K.; Roessler, B.J.; et al. Cloning of cDNAs for human phosphoribosylpyrophosphate synthetases 1 and 2 and X chromosome localization of PRPS1 and PRPS2 genes. Genomics 1990, 8, 555–561.
- Taira, M.; Iizasa, T.; Yamada, K.; Shimada, H.; Tatibana, M. Tissue-differential expression of two distinct genes for phosphoribosyl pyrophosphate synthetase and existence of the testis-specific transcript. Biochim. Biophys. Acta 1989, 1007, 203–208.
- de Brouwer, A.P.M.; Christodoulou, J. Phosphoribosylpyrophosphate snthetase superactivity. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2008; (updated 17 February 2022).
- Arts, W.F.; Loonen, M.C.; Sengers, R.C.; Slooff, J.L. X-linked ataxia, weakness, deafness, and loss of vision in early childhood with a fatal course. Ann. Neurol. 1993, 33, 535–539.
- de Brouwer, A.P.; Williams, K.L.; Duley, J.A.; van Kuilenburg, A.B.; Nabuurs, S.B.; Egmont-Petersen, M.; Lugtenberg, D.; Zoetekouw, L.; Banning, M.J.; Roeffen, M.; et al. Arts syndrome is caused by loss-of-function mutations in PRPS1. Am. J. Hum. Genet. 2007, 81, 507–518.
- Liu, X.; Han, D.; Li, J.; Han, B.; Ouyang, X.; Cheng, J.; Li, X.; Jin, Z.; Wang, Y.; Bitner-Glindzicz, M.; et al. Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2. Am. J. Hum. Genet. 2010, 86, 65–71.
- Liu, X.Z.; Xie, D.; Yuan, H.J.; de Brouwer, A.P.; Christodoulou, J.; Yan, D. Hearing loss and PRPS1 mutations: Wide spectrum of phenotypes and potential therapy. Int. J. Audiol. 2013, 52, 23–28.
- Kim, H.J.; Sohn, K.M.; Shy, M.E.; Krajewski, K.M.; Hwang, M.; Park, J.H.; Jang, S.Y.; Won, H.H.; Choi, B.O.; Hong, S.H.; et al. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (CMTX5). Am. J. Hum. Genet. 2007, 81, 552–558.
- Cunningham, J.T.; Moreno, M.V.; Lodi, A.; Ronen, S.M.; Ruggero, D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 2014, 157, 1088–1103.
- Donini, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.Z.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400.
- Qiu, Z.; Guo, W.; Wang, Q.; Chen, Z.; Huang, S.; Zhao, F.; Yao, M.; Zhao, Y.; He, X. MicroRNA-124 reduces the pentose phosphate pathway and proliferation by targeting PRPS1 and RPIA mRNAs in human colorectal cancer cells. Gastroenterology 2015, 149, 1587–1598.e1511.
- Yang, Y.; Song, L.; Huang, X.; Feng, Y.; Zhang, Y.; Liu, Y.; Li, S.; Zhan, Z.; Zheng, L.; Feng, H.; et al. PRPS1-mediated purine biosynthesis is critical for pluripotent stem cell survival and stemness. Aging 2021, 13, 4063–4078.
- Meng, F.K.; Ellis, T. The second decade of synthetic biology: 2010–2020. Nat. Commun. 2020, 11, 5174.
- Dai, J.B.; Boeke, J.D.; Luo, Z.Q.; Jiang, S.Y.; Cai, Y.Z. Sc3.0: Revamping and minimizing the yeast genome. Genome Biol. 2020, 21, 205.
- Schindler, D. Genetic Engineering and Synthetic Genomics in Yeast to Understand Life and Boost Biotechnology. Bioengineering 2020, 7, 137.
- Wolfe, K.H.; Shields, D.C. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 1997, 387, 708–713.
- Wolfe, K.H. Origin of the Yeast Whole-Genome Duplication. PLoS Biol. 2015, 13, e1002221.
- Botstein, D.; Fink, G.R. Yeast: An experimental organism for 21st Century biology. Genetics 2011, 189, 695–704.
- Burgess, S. Genomics: A matched set of frog sequences. Nature 2016, 538, 320–321.
- Ehrenreich, I.M. Evolution after genome duplication. Science 2020, 368, 1424–1425.
- Schneiter, R.; Carter, A.T.; Hernando, Y.; Zellnig, G.; Schweizer, L.M.; Schweizer, M. The importance of the five phosphoribosyl-pyrophosphate synthetase (Prs) gene products of Saccharomyces cerevisiae in the maintenance of cell integrity and the subcellular localization of Prs1p. Microbiology 2000, 146, 3269–3278.
- Bodenmiller, B.; Wanka, S.; Kraft, C.; Urban, J.; Campbell, D.; Pedrioli, P.G.; Gerrits, B.; Picotti, P.; Lam, H.; Vitek, O.; et al. Phosphoproteomic Analysis Reveals Interconnected System-Wide Responses to Perturbations of Kinases and Phosphatases in Yeast. Sci. Signal. 2010, 3, rs4.
- Stark, C.; Breitkreutz, B.J.; Chatr-Aryamontri, A.; Boucher, L.; Oughtred, R.; Livstone, M.S.; Nixon, J.; Van Auken, K.; Wang, X.; Shi, X.; et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011, 39, D698–D704.
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villen, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682.
- Soulard, A.; Cremonesi, A.; Moes, S.; Schütz, F.; Jenö, P.; Hall, M.N. The Rapamycin-sensitive Phosphoproteome Reveals That TOR Controls Protein Kinase A Toward Some But Not All Substrates. Mol. Biol. Cell 2010, 21, 3475–3486.
- Gnad, F.; de Godoy, L.M.F.; Cox, J.; Neuhauser, N.; Ren, S.; Olsen, J.V.; Mann, M. High-accuracy identification and bioinformatic analysis of in vivo protein phosphorylation sites in yeast. Proteomics 2009, 9, 4642–4652.
- Ficarro, S.B.; McCleland, M.L.; Stukenberg, P.T.; Burke, D.J.; Ross, M.M.; Shabanowitz, J.; Hunt, D.F.; White, F.M. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 2002, 20, 301–305.
- Huber, A.; Bodenmiller, B.; Uotila, A.; Stahl, M.; Wanka, S.; Gerrits, B.; Aebersold, R.; Loewith, R. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009, 23, 1929–1943.
- Loewith, R. A brief history of TOR. Biochem. Soc. Trans. 2011, 39, 437–442.
- Jiménez, A.; Santos, M.A.; Revuelta, J.L. Phosphoribosyl pyrophosphate synthetase activity affects growth and riboflavin production in Ashbya gossypii. BMC Biotechnol. 2008, 8, 67.
- Ugbogu, E.A.; Wippler, S.; Euston, M.; Kouwenhoven, E.N.; de Brouwer, A.P.; Schweizer, L.M.; Schweizer, M. The contribution of the nonhomologous region of Prs1 to the maintenance of cell wall integrity and cell viability. FEMS Yeast Res. 2013, 13, 291–301.
- Herrero, P.; Martinez-Campa, C.; Moreno, F. The hexokinase 2 protein participates in regulatory DNA-protein complexes necessary for glucose repression of the SUC2 gene in Saccharomyces cerevisiae. FEBS Lett. 1998, 434, 71–76.
- Hernando, Y.; Carter, A.T.; Parr, A.; Hove-Jensen, B.; Schweizer, M. Genetic analysis and enzyme activity suggest the existence of more than one minimal functional unit capable of synthesizing phosphoribosyl pyrophosphate in Saccharomyces cerevisiae. J. Biol. Chem. 1999, 274, 12480–12487.
- Hove-Jensen, B. Heterooligomeric phosphoribosyl diphosphate synthase of Saccharomyces cerevisiae: Combinatorial expression of the five PRS genes in Escherichia coli. J. Biol. Chem. 2004, 279, 40345–40350.
- Wang, K.; Vavassori, S.; Schweizer, L.M.; Schweizer, M. Impaired PRPP-synthesizing capacity compromises cell integrity signalling in Saccharomyces cerevisiae. Microbiology 2004, 150, 3327–3339.
- Ugbogu, E.A.; Wang, K.; Schweizer, L.M.; Schweizer, M. Metabolic gene products have evolved to interact with the cell wall integrity pathway in Saccharomyces cerevisiae. FEMS Yeast Res. 2016, 16, fow092.
- Vavassori, S.; Wang, K.; Schweizer, L.M.; Schweizer, M. In Saccharomyces cerevisiae, impaired PRPP synthesis is accompanied by valproate and Li+ sensitivity. Biochem. Soc. Trans. 2005, 33, 1154–1157.
- Vavassori, S. An Investigation of the Influence of Impaired PRPP Production on the Physiology of Saccharomyces cerevisiae. Ph.D. Thesis, Heriot-Watt University, Edinburgh, UK, 2005.
- Wang, K. The Involvement of PRPP Synthetase in Cell Integrity Signalling in Saccharomyces cerevisiae. Ph.D. Thesis, Heriot-Watt University, Edinburgh, UK, 2005.
- Levin, D.E. Cell Wall Integrity Signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2005, 69, 262–291.
- Levin, D.E. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: The cell wall integrity signaling pathway. Genetics 2011, 189, 1145–1175.
- Gonzalez-Rubio, G.; Sellers-Moya, A.; Martin, H.; Molina, M. A walk-through MAPK structure and functionality with the 30-year-old yeast MAPK Slt2. Int. Microbiol. 2021, 24, 531–543.
- Schmitz, H.P.; Lorberg, A.; Heinisch, J.J. Regulation of yeast protein kinase C activity by interaction with the small GTPase Rho1p through its amino-terminal HR1 domain. Mol. Microbiol. 2002, 44, 829–840.
- Heinisch, J.J.; Rodicio, R. Protein kinase C in fungi-more than just cell wall integrity. FEMS Microbiol. Rev. 2018, 42, 22–39.
- Sanz, A.B.; García, R.; Pavón-Vergés, M.; Rodriguez-Peña, J.M.; Arroyo, J. Control of Gene Expression via the Yeast CWI Pathway. Int. J. Mol. Sci. 2022, 23, 1791.
- Jiménez-Gutiérrez, E.; Alegria-Carrasco, E.; Sellers-Moya, A.; Molina, M.; Martin, H. Not just the wall: The other ways to turn the yeast CWI pathway on. Int. Microbiol. 2020, 23, 107–119.
- Stark, M.J.R. Protein phosphorylation and dephosphorylation. pp. 284-375; In The Metabolism and Molecuylar Physiology of Saccharomyces cerevisiae, 2nd ed.; Dickinson, J.R., Schweizer, M., Eds.; CRC Press: Boca Raton, FL, USA; London, UK; New York, NY, USA; Washington, DC, USA, 2004; pp. ix–xv, 1–438.
- Jiménez-Gutiérrez, E.; Alegria-Carrasco, E.; Alonso-Rodriguez, E.; Fernandez-Acero, T.; Molina, M.; Martin, H. Rewiring the yeast cell wall integrity (CWI) pathway through a synthetic positive feedback circuit unveils a novel role for the MAPKKK Ssk2 in CWI pathway activation. FEBS J. 2020, 287, 4881–4901.
- Sauvaget, M.; Hutton, F.; Coull, R.; Vavassori, S.; Wang, K.; Reznik, A.; Chyker, T.; Newfield, C.G.; Euston, E.; Benary, G.; et al. The NHR1-1 of Prs1 and the pentameric motif 284KKCPK288 of Prs3 permit multi-functionality of the PRPP synthetase in Saccharomyces cerevisiae. FEMS Yeast Res. 2019, 19, foz006.
- Ruta, L.L.; Farcasanu, I.C. Saccharomyces cerevisiae and Caffeine Implications on the Eukaryotic Cell. Nutrients 2020, 12, 2440.
- Kuranda, K.; Leberre, V.; Sokol, S.; Palamarczyk, G.; Francois, J. Investigating the caffeine effects in the yeast Saccharomyces cerevisiae brings new insights into the connection between TOR, PKC and Ras/cAMP signalling pathways. Mol. Microbiol. 2006, 61, 1147–1166.
- Loewith, R.; Hall, M.N. Target of Rapamycin (TOR) in Nutrient Signaling and Growth Control. Genetics 2011, 189, 1177–1201.
- Vavassori, S.; Wang, K.; Schweizer, L.M.; Schweizer, M. Ramifications of impaired PRPP synthesis in Saccharomyces cerevisiae. Biochem. Soc. Trans. 2005, 33, 1418–1420.
- Kim, K.Y.; Levin, D.E. Mpk1 MAPK association with the Paf1 complex blocks Sen1-mediated premature transcription termination. Cell 2011, 144, 745–756.
- Costanzo, M.; VanderSluis, B.; Koch, E.N.; Baryshnikova, A.; Pons, C.; Tan, G.; Wang, W.; Usaj, M.; Hanchard, J.; Lee, S.D.; et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 2016, 353, aaf1420.
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795.
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.P.; Muller, J.; Cross, M.J. ERK5: Structure, regulation and function. Cell. Signal. 2012, 24, 2187–2196.
- Truman, A.W.; Millson, S.H.; Nuttall, J.M.; King, V.; Mollapour, M.; Prodromou, C.; Pearl, L.H.; Piper, P.W. Expressed in the yeast Saccharomyces cerevisiae, human ERK5 is a client of the Hsp90 chaperone that complements loss of the Slt2D (Mpk1p) cell integrity stress-activated protein kinase. Eukaryot. Cell 2006, 5, 1914–1924.
- Fasolo, J.; Sboner, A.; Sun, M.G.; Yu, H.; Chen, R.; Sharon, D.; Kim, P.M.; Gerstein, M.; Snyder, M. Diverse protein kinase interactions identified by protein microarrays reveal novel connections between cellular processes. Genes Dev. 2011, 25, 767–778.
- Ugbogu, A.E. The PRS Gene Family Links Primary Metabolism and Cell Signalling in Saccharomyces cerevisiae. Ph.D. Thesis, Heriot-Watt University, Edinburgh, UK, 2014.
- de Brouwer, A.P.; van Bokhoven, H.; Nabuurs, S.B.; Arts, W.F.; Christodoulou, J.; Duley, J. PRPS1 mutations: Four distinct syndromes and potential treatment. Am. J. Hum. Genet. 2010, 86, 506–518.
- Becker, M.A.; Puig, J.G.; Mateos, F.A.; Jimenez, M.L.; Kim, M.; Simmonds, H.A. Inherited superactivity of phosphoribosylpyrophosphate synthetase: Association of uric acid overproduction and sensorineural deafness. Am. J. Med. 1988, 85, 383–390.
- Roessler, B.J.; Golovoy, N.; Palella, T.D.; Heidler, S.; Becker, M.A. Identification of Distinct PRS1 Mutations in Two Patients with X-Linked Phosphoribosylpyrophosphate Synthetase Superactivity. In Purine and Pyrimidine Metabolism in Man VII; Harkness, R.A., Elion, G.B., Zöllner, N., Eds.; Springer: New York, NY, USA, 1991; Volume 309B, pp. 125–128.
- Sperling, O.; Sarapers; Eilam, G.; Devries, A. Accelerated Erythrocyte 5-Phosphoribosyl-1-Pyrophosphate Synthesis. A familial Abnormality Associated with Excessive Uric-Acid Production and Gout. Biochem. Med. 1972, 6, 310–316.
- Taira, M.; Ishijima, S.; Kita, K.; Yamada, K.; Iizasa, T.; Tatibana, M. Nucleotide and deduced amino acid sequences of two distinct cDNAs for rat phosphoribosylpyrophosphate synthetase. J. Biol. Chem. 1987, 262, 14867–14870.
- Iizasa, T.; Taira, M.; Shimada, H.; Ishijima, S.; Tatibana, M. Molecular cloning and sequencing of human cDNA for phosphoribosyl pyrophosphate synthetase subunit II. FEBS Lett. 1989, 244, 47–50.
- Roessler, B.J.; Bell, G.; Heidler, S.; Seino, S.; Becker, M.; Palella, T.D. Cloning of 2 Distinct Copies of Human Phosphoribosylpyrophosphate Synthetase cDNA. Nucleic Acids Res. 1990, 18, 193.
- Ishizuka, T.; Iizasa, T.; Taira, M.; Ishijima, S.; Sonoda, T.; Shimada, H.; Nagatake, N.; Tatibana, M. Promoter Regions of the Human X-Linked Housekeeping Gene-Prps1 and Gene-Prps2 Encoding Phosphoribosylpyrophosphate Synthetase Subunit-I and Subunit-Ii Isoforms. Biochim. Biophys. Acta 1992, 1130, 139–148.
- Sonoda, T.; Taira, M.; Ishijima, S.; Ishizuka, T.; Iizasa, T.; Tatibana, M. Complete nucleotide sequence of human phosphoribosyl pyrophosphate synthetase subunit I (PRS I) cDNA and a comparison with human and rat PRPS gene families. J. Biochem. 1991, 109, 361–364.
- Li, S.; Lu, Y.; Peng, B.; Ding, J. Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. Biochem. J. 2007, 401, 39–47.
- Krath, B.N.; Hove-Jensen, B. Class II recombinant phosphoribosyl diphosphate synthase from spinach. Phosphate independence and diphosphoryl donor specificity. J. Biol. Chem. 2001, 276, 17851–17856.
- Krath, B.N.; Hove-Jensen, B. Implications of secondary structure prediction and amino acid sequence comparison of class I and class II phosphoribosyl diphosphate synthases on catalysis, regulation, and quaternary structure. Protein Sci. 2001, 10, 2317–2324.
- Kadziola, A.; Jepsen, C.H.; Johansson, E.; McGuire, J.; Larsen, S.; Hove-Jensen, B. Novel class III phosphoribosyl diphosphate synthase: Structure and properties of the tetrameric, phosphate-activated, non-allosterically inhibited enzyme from Methanocaldococcus jannaschii. J. Mol. Biol. 2005, 354, 815–828.
- Cherney, M.M.; Cherney, L.T.; Garen, C.R.; James, M.N. The structures of Thermoplasma volcanium phosphoribosyl pyrophosphate synthetase bound to ribose-5-phosphate and ATP analogs. J. Mol. Biol. 2011, 413, 844–856.
- Tang, W.; Li, X.; Zhu, Z.; Tong, S.; Zhang, X.; Teng, M.; Niu, L. Expression, purification, crystallization and preliminary X-ray diffraction analysis of human phosphoribosyl pyrophosphate synthetase 1 (PRS1). Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 432–434.
- Miao, W.; Wang, Y. Targeted Quantitative Kinome Analysis Identifies PRPS2 as a Promoter for Colorectal Cancer Metastasis. J. Proteome Res. 2019, 18, 2279–2286.
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426.
- Katashima, R.; Iwahana, H.; Fujimura, M.; Yamaoka, T.; Itakura, M. Assignment of the human phosphoribosylpyrophosphate synthetase- associated protein 41 gene (PRPSAP2) to 17p11.2-p12. Genomics 1998, 54, 180–181.
- Katashima, R.; Iwahana, H.; Fujimura, M.; Yamaoka, T.; Ishizuka, T.; Tatibana, M.; Itakura, M. Molecular cloning of a human cDNA for the 41-kDa phosphoribosylpyrophosphate synthetase-associated protein. Biochim. Biophys. Acta 1998, 1396, 245–250.
- Ishizuka, T.; Kita, K.; Sonoda, T.; Ishijima, S.; Sawa, K.; Suzuki, N.; Tatibana, M. Cloning and sequencing of human complementary DNA for the phosphoribosylpyrophosphate synthetase-associated protein 39. Biochim. Biophys. Acta 1996, 1306, 27–30.
- Ishizuka, T.; Ahmad, I.; Kita, K.; Sonoda, T.; Ishijima, S.; Sawa, K.; Suzuki, N.; Tatibana, M. The human phosphoribosylpyrophosphate synthetase-associated protein 39 gene (PRPSAP1) is located in the chromosome region 17q24-q25. Genomics 1996, 33, 332–334.
- Sonoda, T.; Ishizuka, T.; Kita, K.; Ishijima, S.; Tatibana, M. Cloning and sequencing of rat cDNA for the 41-kDa phosphoribosylpyrophosphate synthetase-associated protein has a high homology to the catalytic subunits and the 39-kDa associated protein. Biochim. Biophys. Acta 1997, 1350, 6–10.
- Almoguera, B.; He, S.; Corton, M.; Fernandez-San Jose, P.; Blanco-Kelly, F.; Lopez-Molina, M.I.; Garcia-Sandoval, B.; Del Val, J.; Guo, Y.; Tian, L.; et al. Expanding the phenotype of PRPS1 syndromes in females: Neuropathy, hearing loss and retinopathy. Orphanet J. Rare Dis. 2014, 9, 190.
- Synofzik, M.; Muller vom Hagen, J.; Haack, T.B.; Wilhelm, C.; Lindig, T.; Beck-Wodl, S.; Nabuurs, S.B.; van Kuilenburg, A.B.; de Brouwer, A.P.; Schols, L. X-linked Charcot-Marie-Tooth disease, Arts syndrome, and prelingual non-syndromic deafness form a disease continuum: Evidence from a family with a novel PRPS1 mutation. Orphanet J. Rare Dis. 2014, 9, 24.
- Kleineidam, A.; Vavassori, S.; Wang, K.; Schweizer, L.M.; Griac, P.; Schweizer, M. Valproic acid- and lithium-sensitivity in prs mutants of Saccharomyces cerevisiae. Biochem. Soc. Trans. 2009, 37, 1115–1120.
- Kwiatek, J.M.; Gik-Soo, H.; Carman, G.M. Phosphatidate-mediated regulation of lipid synthesis at the nuclear/endoplasmic reticulum membrane. BBA—Mol. Cell. Biol. Lipids 2020, 1865, 15843.
- Duley, J.A.; Christodoulou, J.; de Brouwer, A.P. The PRPP synthetase spectrum: What does it demonstrate about nucleotide syndromes? Nucleosides Nucleotides Nucleic Acids 2011, 30, 1129–1139.
- Zoref, E.; Vries, A.; Sperling, O. Mutant feedback-resistant phosphoribosylpyrophosphate synthetase associated with purine overproduction and gout. Phosphoribosylpyrophosphate and purine metabolism in cultured fibroblasts. J. Clin. Investig. 1975, 56, 1093–1099.
- García-Pavía, P.; Torres, R.J.; Rivero, M.; Ahmed, M.; García-Puig, J.; Becker, M.A. Phosphoribosylpyrophosphate synthetase overactivity as a cause of uric acid overproduction in a young woman. Arthritis Rheum. 2003, 48, 2036–2041.
- Becker, M.A.; Smith, P.R.; Taylor, W.; Mustafi, R.; Switzer, R.L. The genetic and functional basis of purine nucleotide feedback- resistant phosphoribosylpyrophosphate synthetase superactivity. J. Clin. Investig. 1995, 96, 2133–2141.
- Becker, M.A.; Raivio, K.O.; Bakay, B.; Adams, W.B.; Nyhan, W.L. Superactive phosphoribosylpyrophosphate synthetase with altered regulatory and catalytic properties. Adv. Exp. Med. Biol. 1980, 122A, 387–392.
- Becker, M.A.; Losman, M.J.; Wilson, J.; Simmonds, H.A. Superactivity of human phosphoribosyl pyrophosphate synthetase due to altered regulation by nucleotide inhibitors and inorganic phosphate. Biochim. Biophys. Acta 1986, 882, 168–176.
- Becker, M.A.; Taylor, W.; Smith, P.R.; Ahmed, M. Overexpression of the normal phosphoribosylpyrophosphate synthetase 1 isoform underlies catalytic superactivity of human phosphoribosylpyrophosphate synthetase. J. Biol. Chem. 1996, 271, 19894–19899.
- Ahmed, M.; Taylor, W.; Smith, P.R.; Becker, M.A. Accelerated transcription of PRPS1 in X-linked overactivity of normal human phosphoribosylpyrophosphate synthetase. J. Biol. Chem. 1999, 274, 7482–7488.
- Gandía, M.; Fernandez-Toral, J.; Solanellas, J.; Dominguez-Ruiz, M.; Gómez-Rosas, E.; Del Castillo, F.J.; Villamar, M.; Moreno-Pelayo, M.A.; Del Castillo, I. Mutations in PRPS1 causing syndromic or nonsyndromic hearing impairment: Intrafamilial phenotypic variation complicates genetic counseling. Pediatr. Res. 2015, 78, 97–102.
- Moran, R.; Kuilenburg, A.B.; Duley, J.; Nabuurs, S.B.; Retno-Fitri, A.; Christodoulou, J.; Roelofsen, J.; Yntema, H.G.; Friedman, N.R.; van Bokhoven, H.; et al. Phosphoribosylpyrophosphate synthetase superactivity and recurrent infections is caused by a p.Val142Leu mutation in PRS-I. Am. J. Med. Genet. A 2012, 158, 455–460.
- Mittal, R.; Patel, K.; Mittal, J.; Chan, B.; Yan, D.; Grati, M.; Liu, X.Z. Association of PRPS1 Mutations with Disease Phenotypes. Dis. Markers 2015, 2015, 127013.
- Porrmann, J.; Betcheva-Krajcir, E.; Di Donato, N.; Kahlert, A.K.; Schallner, J.; Rump, A.; Schrock, E.; Dobritzsch, D.; Roelofsen, J.; van Kuilenburg, A.B.P.; et al. Novel PRPS1 gain-of-function mutation in a patient with congenital hyperuricemia and facial anomalies. Am. J. Med. Genet. A 2017, 173, 2736–2742.
- Disteche, C.M.; Berletch, J.B. X-chromosome inactivation and escape. J. Genet. 2015, 94, 591–599.
- Balaton, B.P.; Dixon-McDougall, T.; Peeters, S.B.; Brown, C.J. The eXceptional nature of the X chromosome. Hum. Mol. Genet. 2018, 27, R242–R249.
- Yang, B.Y.; Yu, H.X.; Min, J.; Song, X.X. A novel mutation in gene of PRPS1 in a young Chinese woman with X-linked gout: A case report and review of the literature. Clin. Rheumatol. 2020, 39, 949–956.
- Zikánová, M.; Wahezi, D.; Hay, A.; Stiburková, B.; Pitts, C.; Mušálková, D.; Škopová, V.; Barešová, V.; Soucková, O.; Hodanová, K.; et al. Clinical manifestations and molecular aspects of phosphoribosylpyrophosphate synthetase superactivity in females. Rheumatology 2018, 57, 1180–1185.
- Chen, P.; Li, J.; Ma, J.; Teng, M.; Li, X. A small disturbance, but a serious disease: The possible mechanism of D52H-mutant of human PRS1 that causes gout. IUBMB Life 2013, 65, 518–525.
- Maruyama, K.; Ogaya, S.; Kurahashi, N.; Umemura, A.; Yamada, K.; Hashiguchi, A.; Takashima, H.; Torres, R.J.; Aso, K. Arts syndrome with a novel missense mutation in the PRPS1 gene: A case report. Brain Dev. 2016, 38, 954–958.
- de Brouwer, A.P.M.; Christodoulou, J. Arts Syndrome. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2008; (updated 22 March 2018).
- Puusepp, S.; Reinson, K.; Pajusalu, S.; van Kuilenburg, A.B.P.; Dobritzsch, D.; Roelofsen, J.; Stenzel, W.; Õunap, K. Atypical presentation of Arts syndrome due to a novel hemizygous loss-of-function variant in the PRPS1 gene. Mol. Genet. Metab. Rep. 2020, 25, 100677.
- Emmanuel, N.; Ragunathan, S.; Shan, Q.; Wang, F.; Giannakou, A.; Huser, N.; Jin, G.X.; Myers, J.; Abraham, R.T.; Unsal-Kacmaz, K. Purine Nucleotide Availability Regulates mTORC1 Activity through the Rheb GTPase. Cell Rep. 2017, 19, 2665–2680.
- Lenherr, N.; Christodoulou, J.; Duley, J.; Dobritzsch, D.; Fairbanks, L.; Datta, A.N.; Filges, I.; Gurtler, N.; Roelofsen, J.; van Kuilenburg, A.B.P.; et al. Co-therapy with S-adenosylmethionine and nicotinamide riboside improves t-cell survival and function in Arts Syndrome (PRPS1 deficiency). Mol. Genet. Metab. Rep. 2021, 26, 100709.
- Bottiglieri, T. S-Adenosyl-L-methionine (SAMe): From the bench to the bedside--molecular basis of a pleiotrophic molecule. Am. J. Clin. Nutr. 2002, 76, 1151S–1157S.
- Chi, Y.; Sauve, A.A. Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 657–661.
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547.
- Liu, R.; Li, J.Y.; Shao, J.C.; Lee, J.H.; Qiu, X.M.; Xiao, Y.X.; Zhang, B.W.; Hao, Y.L.; Li, M.; Chen, Q.M. Innate immune response orchestrates phosphoribosyl pyrophosphate synthetases to support DNA repair. Cell Metab. 2021, 33, 2076–2089.e9.
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55.
- Mullighan, C.G. Mutant PRPS1: A new therapeutic target in relapsed acute lymphoblastic leukemia. Nat. Med. 2015, 21, 553–554.
- Li, T.; Song, L.; Zhang, Y.; Han, Y.; Zhan, Z.; Xv, Z.; Li, Y.; Tang, Y.; Yang, Y.; Wang, S.; et al. Molecular mechanism of c-Myc and PRPS1/2 against thiopurine resistance in Burkitt’s lymphoma. J. Cell. Mol. Med. 2020, 24, 6704–6715.
- Li, B.; Li, H.; Bai, Y.; Kirschner-Schwabe, R.; Yang, J.J.; Chen, Y.; Lu, G.; Tzoneva, G.; Ma, X.; Wu, T.; et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat. Med. 2015, 21, 563–571.
- Xue, C.; Yu, D.M.; Gherardi, S.; Koach, J.; Milazzo, G.; Gamble, L.; Liu, B.; Valli, E.; Russell, A.J.; London, W.B.; et al. MYCN promotes neuroblastoma malignancy by establishing a regulatory circuit with transcription factor AP4. Oncotarget 2016, 7, 54937–54951.
- Hsieh, A.C.; Ruggero, D. Targeting Eukaryotic Translation Initiation Factor 4E (eIF4E) in Cancer. Clin. Cancer Res. 2010, 16, 4914–4920.
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61.
- Hsieh, A.L.; Walton, Z.E.; Altman, B.J.; Stine, Z.E.; Dang, C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21.
- Lei, B.; Xie, L.X.; Zhang, S.B.; Wan, B.; Zhong, L.R.; Zhou, X.M.; Mao, X.M.; Shu, F.P. Phosphoribosyl-pyrophosphate synthetase 2 (PRPS2) depletion regulates spermatogenic cell apoptosis and is correlated with hypospermatogenesis. Asian J. Androl. 2020, 22, 493–499.
- Luo, Y.; Yuan, J.; Huang, J.; Yang, T.; Liu, M.; Chen, J.; Huang, W.; Zhang, H. Role of PRPS2 as a prognostic and therapeutic target in osteosarcoma. J. Clin. Pathol. 2021, 74, E12.
- Ma, Y.; An, X.; Guan, X.; Kong, Q.; Wang, Y.; Li, P.; Meng, Y.; Cui, Y.; Wen, X.; Guo, Y.; et al. High expression of PRPS1 induces an anti-apoptotic effect in B-ALL cell lines and predicts an adverse prognosis in Chinese children with B-ALL. Oncol. Lett. 2018, 15, 4314–4322.
- He, M.; Chao, L.; You, Y.P. PRPS1 silencing reverses cisplatin resistance in human breast cancer cells. Biochem. Cell Biol. 2017, 95, 385–393.
- Minchenko, O.H.; Garmash, I.A.; Kovalevska, O.V.; Tsymbal, D.O.; Minchenko, D.O. Expression of phosphoribosyl pyrophosphate synthetase genes in U87 glioma cells with ERN1 knockdown: Effect of hypoxia and endoplasmic reticulum stress. Ukr. Biochem. J. 2014, 86, 74–83.
- Zhu, Y.; Li, T.; Ramos da Silva, S.; Lee, J.J.; Lu, C.; Eoh, H.; Jung, J.U.; Gao, S.J. A Critical Role of Glutamine and Asparagine γ-Nitrogen in Nucleotide Biosynthesis in Cancer Cells Hijacked by an Oncogenic Virus. mBio 2017, 8, e01179-17.
- Angius, A.; Uva, P.; Pira, G.; Muroni, M.R.; Sotgiu, G.; Saderi, L.; Uleri, E.; Caocci, M.; Ibba, G.; Cesaraccio, M.R.; et al. Integrated Analysis of miRNA and mRNA Endorses a Twenty miRNAs Signature for Colorectal Carcinoma. Int. J. Mol. Sci. 2019, 20, 4067.
- Agrahari, A.K.; Sneha, P.; George Priya Doss, C.; Siva, R.; Zayed, H. A profound computational study to prioritize the disease-causing mutations in PRPS1 gene. Metab. Brain Dis. 2018, 33, 589–600.
- Preethi, B.; Ramanathan, K. In silico analysis of functional nsSNPs of the human PRPS1 gene. Res. J. Pharm. Biol. Chem. Sci. 2015, 6, 846–851.
- Li, X.; Qian, X.; Peng, L.X.; Jiang, Y.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Lee, J.H.; Cote, G.; Wang, H.; et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat. Cell Biol. 2016, 18, 561–571.
- Yang, J.; Yang, S.; Wang, Q.; Pang, J.; Wang, Y.; Wang, H.; Fu, X. KHK-A promotes the proliferation of oesophageal squamous cell carcinoma through the up-regulation of PRPS1. Arab J. Gastroenterol. 2021, 22, 40–46.
- Li, C.; Yan, Z.; Cao, X.; Zhang, X.; Yang, L. Phosphoribosylpyrophosphate Synthetase 1 Knockdown Suppresses Tumor Formation of Glioma CD133+ Cells Through Upregulating Cell Apoptosis. J. Mol. Neurosci. 2016, 60, 145–156.
- Yang, L.; Yan, Z.; Wang, Y.; Ma, W.; Li, C. Down-expression of miR-154 suppresses tumourigenesis in CD133(+) glioblastoma stem cells. Cell Biochem. Funct. 2016, 34, 404–413.
- Li, J.; Ye, J.; Zhu, S.; Cui, H. Down-Regulation of Phosphoribosyl Pyrophosphate Synthetase 1 Inhibits Neuroblastoma Cell Proliferation. Cells 2019, 8, 955.
- Jing, X.; Wang, X.J.; Zhang, T.; Zhu, W.; Fang, Y.; Wu, H.; Liu, X.; Ma, D.; Ji, X.; Jiang, Y.; et al. Cell-Cycle-Dependent Phosphorylation of PRPS1 Fuels Nucleotide Synthesis and Promotes Tumorigenesis. Cancer Res. 2019, 79, 4650–4664.
- Zhang, F.; Patel, D.M.; Colavita, K.; Rodionova, I.; Buckley, B.; Scott, D.; Kumar, A.; Shabalina, S.A.; Saha, S.; Chernov, M.; et al. Arginylation regulates purine biosynthesis by enhancing the biological activity of phosphoribosyl pyrophospate synthase. Nat. Comm. 2015, 6, 7517.
- Kaida, A.; Ariumi, Y.; Baba, K.; Matsubae, M.; Takao, T.; Shimotohno, K. Identification of a novel p300-specific-associating protein, PRS1 (phosphoribosylpyrophosphate synthetase subunit 1). Biochem. J. 2005, 391, 239–247.
- Qiao, H.; Tan, X.; Lv, D.J.; Xing, R.W.; Shu, F.P.; Zhong, C.F.; Li, C.; Zou, Y.G.; Mao, X.M. Phosphoribosyl pyrophosphate synthetases 2 knockdown inhibits prostate cancer progression by suppressing cell cycle and inducing cell apoptosis. J. Cancer 2020, 11, 1027–1037.
- Qian, X.; Li, X.; Tan, L.; Lee, J.H.; Xia, Y.; Cai, Q.; Zheng, Y.; Wang, H.; Lorenzi, P.L.; Lu, Z. Conversion of PRPS Hexamer to Monomer by AMPK-Mediated Phosphorylation Inhibits Nucleotide Synthesis in Response to Energy Stress. Cancer Discov. 2018, 8, 94–107.
- Lei, B.; Wan, B.; Peng, J.; Yang, Y.; Lv, D.; Zhou, X.; Shu, F.; Li, F.; Zhong, L.; Wu, H.; et al. PRPS2 Expression Correlates with Sertoli-Cell Only Syndrome and Inhibits the Apoptosis of TM4 Sertoli Cells. J. Urol. 2015, 194, 1491–1497.
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 20, 661–673.
- Wang, D.; Chen, Y.; Fang, H.; Zheng, L.; Li, Y.; Yang, F.; Xu, Y.; Du, L.; Zhou, B.S.; Li, H. Increase of PRPP enhances chemosensitivity of PRPS1 mutant acute lymphoblastic leukemia cells to 5-Fluorouracil. J. Cell. Mol. Med. 2018, 22, 6202–6212.