REGγ, a proteasome activator belonging to the 11S (otherwise known as REG, PA28, or PSME) proteasome activator family, is widely present in many eukaryotes. By binding to the 20S catalytic core particle, REGγ acts as a molecular sieve to selectively target proteins for degradation in an ATP- and ubiquitin-independent manner. This non-canonical proteasome pathway directly regulates seemingly unrelated cellular processes including cell growth and proliferation, apoptosis, DNA damage response, immune response, and metabolism. By affecting different pathways, REGγ plays a vital role in the regulation of cellular life and death through the maintenance of protein homeostasis.
- REGγ
- proteasome
- tumor suppressor
- regulation
1. Introduction
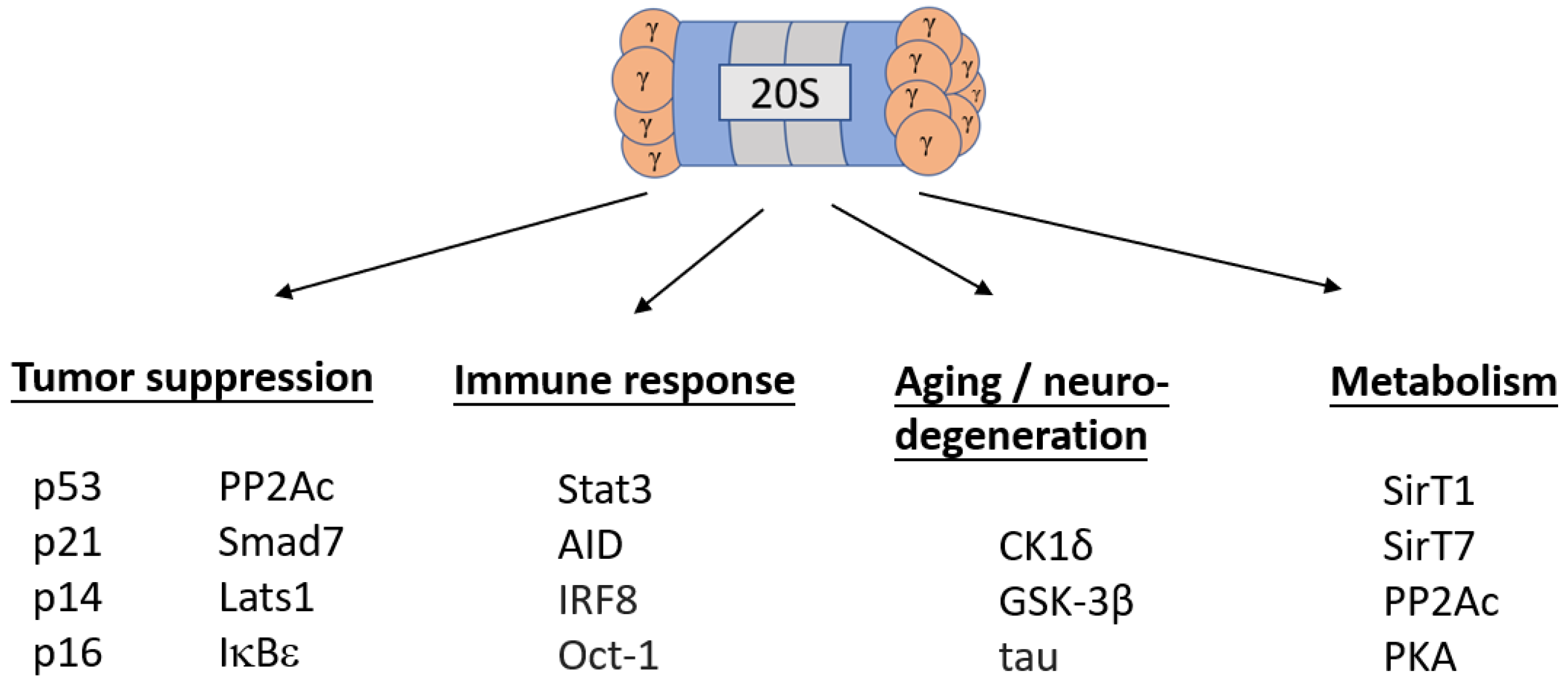
2. Regulation of Life
2.1. Cell Growth and Proliferation
2.2. Energy Metabolism
2.3. Reproduction
3. Regulation of Death
In contrast to its role as a regulator of various processes involved in maintaining cell life, REGγ also contributes to cell death upon its loss of function. As shown previously, overexpression of REGγ often contributes to tumor formation and excessive growth, but underexpression has also been linked with increased levels of apoptosis, aging and neurodegenerative disorders, reduced spindle integrity, and slower DNA damage repair.
3.1. Apoptosis
One of the most notable functions of REGγ as it relates to the regulation of death is its capacity to cause apoptosis upon attenuation. Several studies have found a correlation between reduced levels of REGγ and increased levels of apoptosis [15,25,38–40]. The fact that depletion of REGγ sensitized cells to stress-induced apoptosis is likely due to the deregulation of p53 [41]. Mechanistically, p53 is known to stimulate apoptosis via transcriptional upregulation of pro-apoptotic proteins PUMA and NOXA [42]. Thus, reduction of REGγ contributes to increased levels of p53 as well as the pro-apoptotic factors that lead to apoptosis. Additionally, REGγ is a known substrate of caspases 3 and 7, so these caspases attenuate REGγ and further reinforce apoptosis. Interestingly, REGγ proteins inhibited caspase activity in vitro, indicating a mutually inhibitive relationship [43]. Thus, a loss of REGγ could upregulate apoptosis due to a lack of caspase inhibition. Furthermore, in a study on starvation-induced proteasome assemblies, inhibition of either REGγ or RAD23B, a proteasome shuttling factor, in amino acid-depleted cells prevented p53/NOXA upregulation and apoptosis [44]. This seems to indicate that the down-regulatory effect that REGγ has on apoptosis under normal conditions is reversed in times of cellular energy deficit. This data coincides with REGγ’s role in preserving cellular energy levels during starvation since the REGγ-proteasome appears to contribute to tissue fitness under such conditions. Hence, through the regulation of p53 and inhibition of caspase activity, REGγ is capable of inhibiting apoptosis.
3.2. Aging & Neurodegenerative Disease
Recent studies have indicated a relationship between REGγ and both aging and neurodegenerative disease, particularly in conditions of REGγ deficiency/decline. Firstly, REGγ deficiency was found to cause premature aging in mice via the REGγ-CK1δ-Mdm2-p53 pathway, whereby lower levels of REGγ were associated with accumulation of CK1δ and p53, leading to premature aging [10]. Consistently, an RNA-seq comparison analysis between 40- and 70-year-old human cortexes showed that REGγ expression was reduced in aged human beings [45]. Interestingly, a microarray analysis of hippocampal CA1 regions from 31 postmortem AD patients found a nearly 4-fold reduction in REGγ expression compared to normal controls [46]. These data indicate not only that REGγ expression diminishes as individuals age, but also that this decrease may be linked with neurodegenerative disorders. Providing a potential explanation for the correlation between REGγ decline and brain disorders, researchers showed that REGγ knockout mice exhibited increased GSK-3β activity and experienced several cognitive deficiencies such as defective prepulse inhibition (PPI), decreased working memory, and disability in nest building. Since GSK-3β overexpression has been linked with CNS diseases such as schizophrenia, REGγ-mediated regulation of GSK-3β is a likely mechanism through which REGγ affects the CNS [24]. Consistently, inhibition of GSK-3β was sufficient to rescue the compromised PPI phenotypes and deficiency in working memory. In another study of Huntington’s Disease (HD), patients were found to have reduced proteolytic activity in the brain and other tissues, leading to intraneuronal nuclear protein aggregates of mutant huntingtin. Interestingly, overexpression of REGγ was sufficient to rescue proteasome function in HD cells. At the same time, REGγ could improve cell viability in mutant-huntingtin expressing striatal neurons in the presence of pathological stressors such as quinolinic acid and MG132, a reversible proteasome inhibitor [47]. Similarly, a later study found that injecting lenti-REGγ virus into the striatum of mutant huntingtin-expressing mice improved motor control and helped to restore proteolytic activity to the UPS [48]. Thus, a decline in REGγ can contribute to both aging and neurodegenerative disorders via the pathways that lead to the accumulation of p53, tau, CK1δ, and GSK-3β.
3.3. DNA Damage Repair & Chromosomal Stability
REGγ is also an important factor in both DNA damage repair and chromosomal stability, such that its deficiency could lead to an increased likelihood of cell death during or after cell division due to slower repair of DNA damage, defective spindle structures, and aneuploidy. Firstly, REGγ is involved in double strand break (DSB) repair, as reduced REGγ levels result in longer repair times and more DNA damage hallmarks in several human cell lines. Mechanistically, REGγ was found to be rapidly localized to the site of DNA damage by ataxia-telangiectasia mutated (ATM) protein kinase and to recruit the 20S proteasome to the damaged site for efficient repair [49]. Supporting these results, a recent study on the involvement of lens epithelium-derived growth factor (LEDGF/p75), a transcriptional coactivator involved in DSB repair, on DNA damage repair found that LEDGF-depleted cells exhibit decreased REGγ protein levels and persistent activation of DNA damage signals such as γH2AX and BRCA1, indicating that the REGγ-proteasome likely plays a role in degrading molecules involved in DNA damage response [50]. Additionally, REGγ has been found to localize on chromosomes during the telophase and regulate spindle integrity, independent of the 20S proteasome. In this study, when cells were treated with the spindle damaging agent nocodazole, REGγ overexpression weakened mitotic arrest to trigger premature exit from mitosis, whereas REGγ underexpression exhibited the opposite effect. Furthermore, REGγ (−/−) mice and human fibroblasts with depleted expression of REGγ exhibited a marked aneuploidy and an increased frequency of abnormal metaphases, suggesting REGγ’s role in maintaining chromosomal stability [6]. In addition to its role in regulating chromosomal stability during mitosis, REGγ also appears to control the compaction of chromatin in a manner not dependent on binding with the 20S proteasome. In this study on a human cell line, FLIM-FRET microscopy analysis revealed that REGγ depletion correlates with chromatin decompaction, likely through REGγ-mediated maintenance of histone modifications H3K9me3 and H4K20me3 [7]. As such, REGγ is capable of regulating DNA damage repair, mitotic spindle integrity, and chromosomal compaction to maintain genomic stability.
Thus, based on loss-of-function studies, REGγ deficiency leads to cell death through various pathways. Firstly, REGγ prevents apoptosis through the downregulation of p53 and caspase such that its deficiency most likely leads to cell death. Furthermore, reduction of REGγ levels such as that which occurs with aging greatly facilitates p53/CK1δ/tau accumulation and GSK-3β overexpression, further leading to neurodegenerative disorders. In addition, REGγ deficiency leads to chromosomal instability and generates significant defects in DNA damage repair, which markedly reduces the viability of both cells and organisms. Therefore, REGγ is a key regulator of cell death processes.
4. Regulation of the Regulator
The evidence described above establishes REGγ as a regulator of various processes involved in both the life and death of cells. To carry out its specific functions, the expression and distribution of REGγ are manipulated at various levels, which has been revealed in the recent studies shown henceforth.
4.1. Transcriptional Regulation
As it relates to the transcriptional regulation of REGγ, p53/TGF-β signaling has been found to inhibit REGγ expression by Smad-dependent interaction with the REGγ promoter region. In this form of repression, p53 binds to the p53 response element (p53RE), a DNA binding domain in the REGγ promoter region. The activated TGF-β pathway triggers a p53-Smad3 inhibitory complex, followed by the formation of a Smad3/Nuclear receptor co-repressor 1 (N-CoR) complex for the repression of REGγ transcription. Mutant p53 was still able to bind to the p53RE region but prevented the formation of the Smad3/N-CoR complex. By doing so, mutant p53 can enhance the transcription of REGγ via prevention of inhibition, thus acting as an oncogene and contributing to cancer development [51]. In another study on endometrial cancer (EC), mutant p53-R248Q, the second most common p53 ‘hot spot’ mutation, was found to upregulate the expression of REGγ, as increased levels of mutant p53 correlated with increased levels of REGγ. Ultimately, this mutant p53-REGγ oncogenic pathway contributed to the evolution of EC [52]. Additionally, a genomic sequence analysis of a Drosophila cell line revealed that the REGγ promoter region contains transcription regulatory elements which often appear in the promoters of DNA replication or cell cycle progression genes. This indicates that expression of cell cycle regulatory proteins would be correlated with REGγ expression [27]. Thus, REGγ transcription is regulated by the p53/TGF-β signaling cascade and other cell cycle-regulatory factors.
4.2. Post-transcriptional Regulation
At the post-transcriptional level, miR-7-5p was found to bind to the REGγ 3’UTR for reduction of both mRNA and protein levels. In a breast cancer cell line, activation of miR-7-5p signaling inhibits cell proliferation, leading to apoptosis. Expectedly, introduction of an miR-7-5p inhibitor resulted in increased REGγ protein levels [43], and Cerebellar degeneration-related protein 1 antisense RNA (CDR1as), a circular RNA involved in the inhibition of miR-7, led to upregulation of REGγ in a breast cancer cell line [53]. Confirming these results, a study on gastric cancer cells found that downregulation of CDR1as promoted the cytotoxic effects of a traditional cancer drug Diosbulbin-B by inhibiting REGγ via the upregulation of miR-7-5p [54]. Similarly, miR-195-5p was also found to inhibit REGγ, as an miR-195-5p inhibitor prevents apoptosis and increases cell growth by preventing the inhibition of REGγ [40]. Therefore, REGγ is downregulated at the post-transcriptional level via miRNA interference.
4.3. Post-translational Regulation
Interestingly, a recent study found that NEFA-interacting nuclear protein 30 (NIP30), a negative regulator of REGγ, binds directly to REGγ for its inhibition. Activation of NIP30 attenuates cancer cell growth and sensitizes p53-compromised cells to chemotherapy. The study also showed that p21 protein levels were upregulated in the presence of NIP30 but p21 mRNA levels were unaffected, indicating REGγ’s inability to degrade p21 in the presence of NIP30. Cell division cycle 25A (CDC25A), a key cell cycle regulatory phosphatase that is degraded in response to DNA damage, was found to dephosphorylate NIP30 for its inactivation, thus preventing it from binding to REGγ. DNA damage by UV radiation reduced CDC25A levels sharply, which results in increased NIP30 phosphorylation, leading to inhibition of p21 degradation. Thus, a CDC25A-NIP30-REGγ regulatory axis can be established [55]. Similarly, another recent study on proteasomal inhibition in multiple myeloma (MM) cells found that indirubine-3’-monoxime (I3MO), a derivative of indirubin, significantly suppresses the growth of MM cells by directly binding to and inhibiting REGγ. This study also showed that cells resistant to bortezomib, an inhibitor of the 20S catalytic core particle, could be sensitized to bortezomib-induced apoptosis upon introduction of I3MO and subsequent REGγ inhibition [56].
In addition to regulation by NIP30 and I3MO, REGγ can be SUMOylated at multiple sites by SUMO-1, SUMO-2, and SUMO-3 to control its distribution and stability in the cell. In SUMOylation-deficient cells, REGγ was found to have an attenuated ability to degrade p21, suggesting the functional role of SUMOylation for both improving REGγ-mediated degradation and allowing it to target a broader range of substrates [57]. Additionally, it was found that the REGγ-proteasome complex degrades proteins such as p21 and HCV core protein more rapidly in an oxidative environment, and antioxidants counteract this oxidation-induced protein degradation. The addition of MG132, a proteasome inhibitor, or silencing of REGγ were both able to block this oxidant-induced degradation of p21. Hence, REGγ activity can be regulated through the manipulation of the oxidative state of the surrounding cellular environment [58]. In summary, REGγ can be regulated post-transcriptionally by manipulation of NIP30, CDC25A, SUMOylation, and modification of the oxidative state of the cellular environment to control the degradative ability of the REGγ-proteasome complex.
REGγ is transcriptionally regulated by p53-Smad3-dependent repression, mutant p53-mediated upregulation, and transcription factors functioning in cell cycle progression. After transcription, miR-7-5p and miR-195-5p are able to target REGγ for inhibition, and the addition of miRNA inhibitors such as CDR1as can prevent this interference. Lastly, REGγ can be regulated post-translationally with SUMOylation, NIP30 binding, CDC25A-mediated deactivation of NIP30, or alteration of the oxidative state of the cellular environment to control the distribution of REGγ and the degradative capacity of the REGγ-proteasome complex.
5. Conclusion and Future Directions
As shown previously, REGγ plays a decisive role in maintaining the homeostasis of the cellular proteome (Figure 2). Given the facts that REGγ regulates cellular health in a plethora of ways and that mutations in REGγ are rarely discovered, dysregulation affecting the expression of REGγ inevitably carries significant consequences for both cellular and organismal well-being.
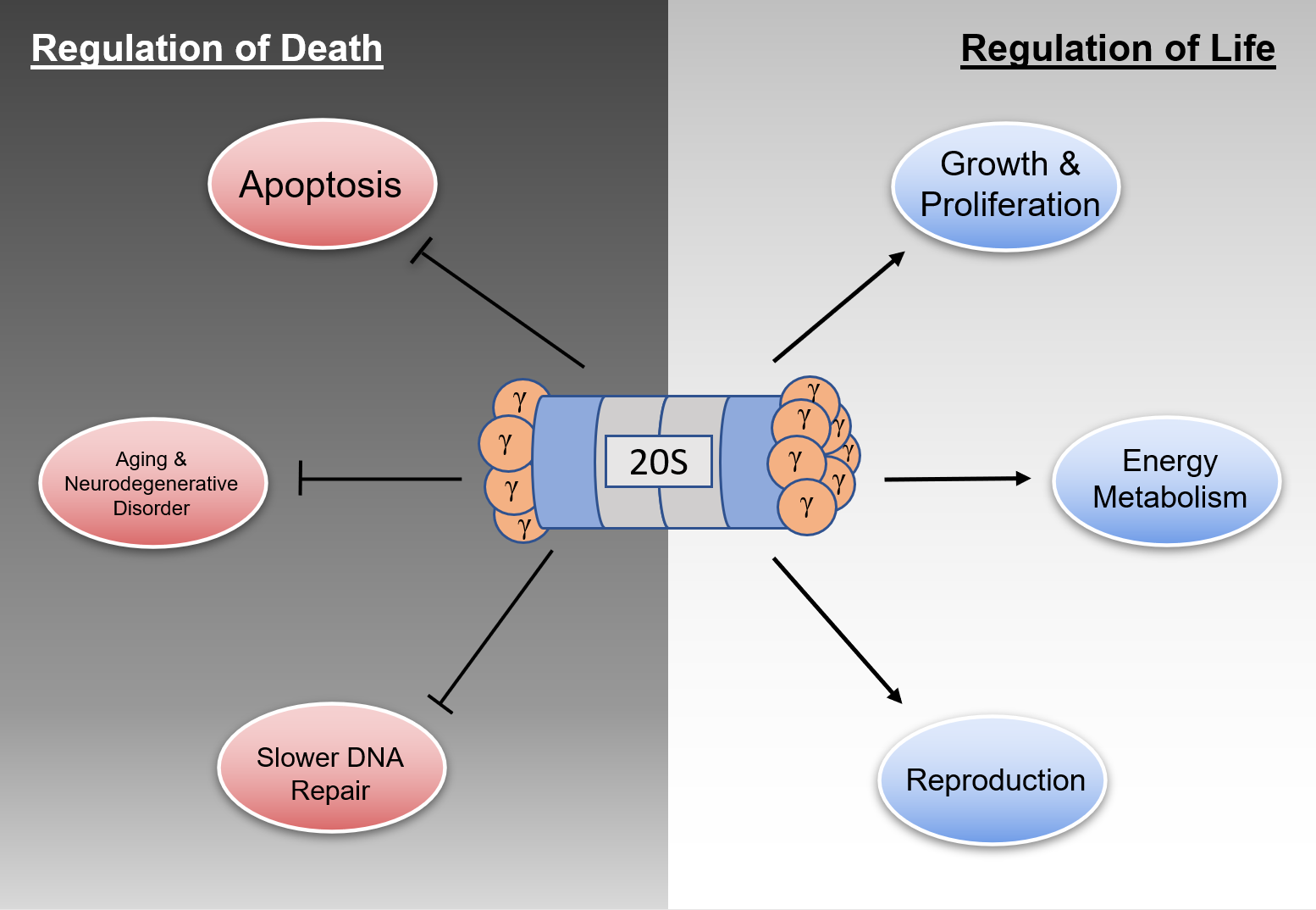
Figure 2. Regulatory functions of the REGγ-proteasome. Under normal expression conditions, REGγ maintains homeostasis via maintenance of processes listed under Regulation of Life and downregulation of processes listed under Regulation of Death.
With research having uncovered various new pathways for the regulation of REGγ at the transcriptional, post-transcriptional, and post-translational levels, manipulation of REGγ expression via therapeutic targeting becomes a viable and important direction for future research. Despite numerous studies, data regarding both the structure and substrate targeting mechanisms of REGγ are still lacking. In order to develop a foundation of knowledge for future studies, it will be necessary to first conduct structural analysis of the REGγ-proteasome in complexes with substrates in order to reveal the mechanistic details surrounding substrate binding and degradation. These data could help to elucidate the specific chemical and physical properties that facilitate REGγ-mediated protein degradation. Such studies focusing on the mechanism of substrate recognition by REGγ will facilitate the discovery of additional substrates and their functions. Furthermore, with research having already identified REGγ as a potential drug target by using NIP30 inhibition, crystallographic and electron microscopic analysis of the REGγ-proteasome-NIP30 complex could reveal new inhibitory mechanisms. Given this information, new inhibitory compounds could be engineered to safely treat patients with drug-resistant cancer.This entry is adapted from the peer-reviewed paper 10.3390/cells11152281
References
- Hochstrasser, M. Ubiquitin, Proteasomes, and the Regulation of Intracellular Degradation. Curr. Opin. Cell Biol. 1995, 7, 215–223. https://doi.org/10.1016/0955-0674(95)80031-x.
- Tanaka, K. The Proteasome: Overview of Structure and Functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. https://doi.org/10.2183/pjab.85.12.
- Cascio, P. PA28γ: New Insights on an Ancient Proteasome Activator. Biomolecules 2021, 11, 228. https://doi.org/10.3390/biom11020228.
- Li, J.; Rechsteiner, M. Molecular Dissection of the 11S REG (PA28) Proteasome. Biochimie 2001, 83, 373–383. https://doi.org/10.1016/s0300-9084(01)01236-6.
- Wójcik, C.; Tanaka, K.; Paweletz, N.; Naab, U.; Wilk, S. Proteasome Activator (PA28) Subunits, α, β and γ (Ki Antigen) in NT2 Neuronal Precursor Cells and HeLa S3 Cells. Eur. J. Cell Biol. 1998, 77, 151–160. https://doi.org/10.1016/S0171-9335(98)80083-6.
- Zannini, L.; Lecis, D.; Buscemi, G.; Carlessi, L.; Gasparini, P.; Fontanella Enrico and Lisanti, S.; Barton, L.; Delia, D. REGgamma Proteasome Activator Is Involved in the maintenance of Chromosomal Stability. Cell Cycle 2008, 7, 504–512. https://doi.org/10.4161/cc.7.4.5355.
- Fesquet, D.; Llères, D.; Grimaud, C.; Viganò, C.; Méchali, F.; Boulon, S.; Coux, O.; Bonne-Andrea, C.; Baldin, V. The 20S Proteasome Activator PA28γ Controls the compaction of Chromatin. J. Cell Sci. 2021, 134, jcs257717. https://doi.org/10.1242/jcs.257717.
- Fabre, B.; Lambour, T.; Garrigues, L.; Ducoux-Petit, M.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.-P. Label-Free Quantitative Proteomics Reveals the Dynamics of Proteasome Complexes Composition and Stoichiometry in a Wide Range of Human Cell Lines. J. Proteome Res. 2014, 13, 3027–3037. https://doi.org/10.1021/pr500193k.
- Li, X.; Lonard, D.M.; Jung, S.Y.; Malovannaya, A.; Feng, Q.; Qin, J.; Tsai Sophia, Y.; Tsai, M.-J.; O’Malley, B.W. The SRC-3/AIB1 Coactivator Is Degraded in a Ubiquitin- and ATP-Independent Manner by the REGgamma Proteasome. Cell 2006, 124, 381–392. https://doi.org/10.1016/j.cell.2005.11.037.
- Li, L.; Zhao, D.; Wei, H.; Yao, L.; Dang, Y.; Amjad, A.; Xu, J.; Liu, J.; Guo, L.; Li, D.; et al. REGγ Deficiency Promotes Premature Aging via the Casein 1 Pathway. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11005–11010. https://doi.org/10.1073/pnas.1308497110.
- Li, X.; Amazit, L.; Long, W.; Lonard, D.M.; Monaco, J.J.; O’Malley, B.W. Ubiquitin- and ATP-Independent Proteolytic Turnover of P21 by the REGgamma-Proteasome Pathway. Mol. Cell 2007, 26, 831–842. https://doi.org/10.1016/j.molcel.2007.05.028.
- Chen, X.; Barton, L.F.; Chi, Y.; Clurman, B.E.; Roberts, J.M. Ubiquitin-Independent Degradation of Cell-Cycle Inhibitors by the REGgamma Proteasome. Mol. Cell 2007, 26, 843–852. https://doi.org/10.1016/j.molcel.2007.05.022.
- Wang, Q.; Gao, X.; Yu, T.; Yuan, L.; Dai, J.; Wang, W.; Chen, G.; Jiao, C.; Zhou, W.; Huang, Q.; et al. REGγ Controls Hippo Signaling and Reciprocal-ΚB–YAP Regulation to Promote Colon Cancer. Clin. Cancer Res. 2018, 24, 2015–2025. https://doi.org/10.1158/1078-0432.CCR-17-2986.
- Yao, L.; Zhou, L.; Xuan, Y.; Zhang, P.; Wang, X.; Wang, T.; Meng, T.; Xue, Y.; Ma, X.; Shah, A.S.; et al. The Proteasome Activator REGγ Counteracts Expression and Autoimmunity. J. Autoimmun. 2019, 103, 102282. https://doi.org/10.1016/j.jaut.2019.05.010.
- Liu, S.; Zheng, L.-L.; Zhu, Y.-M.; Shen, H.-J.; Zhong, Q.; Huang, J.; Li, C.; Liu Zhi and Yao, M.-D.; Ou, R.-M.; Zhang, Q. Knockdown of REGγ Inhibits the Proliferation and migration and Promotes the Apoptosis of Multiple Myeloma Cells by downregulating NF-ΚB Signal Pathway. Hematology 2018, 23, 277–283. https://doi.org/10.1080/10245332.2017.1385194.
- Jiao, C.; Li, L.; Zhang, P.; Zhang, L.; Li, K.; Fang, R.; Yuan, L.; Shi, K.; Pan, L.; Guo, Q.; et al. REGγ Ablation Impedes Dedifferentiation of Anaplastic Carcinoma and Accentuates Radio-Therapeutic Response by regulating the Smad7-TGF-β Pathway. Cell Death Differ. 2020, 27, 497–508. https://doi.org/10.1038/s41418-019-0367-9.
- Tong, L.; Shen, S.; Huang, Q.; Fu, J.; Wang, T.; Pan, L.; Zhang, P.; Chen, G.; Huang, T.; Li, K.; et al. Proteasome-Dependent Degradation of Smad7 Is Critical for Lung Metastasis. Cell Death Differ. 2020, 27, 1795–1806. https://doi.org/10.1038/s41418-019-0459-6.
- Uchimura, Y.; Barton, L.F.; Rada, C.; Neuberger, M.S. REG-γ Associates with and Modulates the Abundance of nuclear Activation-Induced Deaminase. J. Exp. Med. 2011, 208, 2385–2391. https://doi.org/10.1084/jem.20110856.
- Zhou, L.; Yao, L.; Zhang, Q.; Xie, W.; Wang, X.; Zhang, H.; Xu, J.; Lin, Q.; Li, Q.; Xuan, Y.; et al. REGγ Controls Th17 Cell Differentiation and Autoimmune by Regulating Dendritic Cells. Cell. Mol. Immunol. 2020, 17, 1136–1147. https://doi.org/10.1038/s41423-019-0287-0.
- Fan, J.; Liu, L.; Liu, Q.; Cui, Y.; Yao, B.; Zhang, M.; Gao, Y.; Fu, Y.; Dai, H.; Pan, J.; et al. CKIP-1 Limits Foam Cell Formation and Inhibits atherosclerosis by Promoting Degradation of Oct-1 by REGγ. Nat. Commun. 2019, 10, 425. https://doi.org/10.1038/s41467-018-07895-3.
- Dong, S.; Jia, C.; Zhang, S.; Fan, G.; Li, Y.; Shan, P.; Sun, L.; Xiao, W.; Li, L.; Zheng, Y.; et al. The REGγ Proteasome Regulates Hepatic Lipid metabolism through Inhibition of Autophagy. Cell Metab. 2013, 18, 380–391. https://doi.org/10.1016/j.cmet.2013.08.012.
- Sun, L.; Fan, G.; Shan, P.; Qiu, X.; Dong, S.; Liao, L.; Yu, C.; Wang, T.; Gu, X.; Li, Q.; et al. Regulation of Energy Homeostasis by the Ubiquitin-Independent Proteasome. Nat. Commun. 2016, 7, 12497. https://doi.org/10.1038/ncomms12497.
- Liu, S.; Lai, L.; Zuo, Q.; Dai, F.; Wu, L.; Wang, Y.; Zhou, Q.; Liu, J.; Liu, J.; Li, L.; et al. PKA Turnover by the REGγ-Proteasome Modulates FoxO1 Activity and VEGF-Induced Angiogenesis. J. Mol. Cell. Cardiol. 2014, 72, 28–38. https://doi.org/10.1016/j.yjmcc.2014.02.007.
- Lv, Y.; Meng, B.; Dong, H.; Jing, T.; Wu, N.; Yang, Y.; Huang, L.; Moses, R.E.; O’Malley, B.W.; Mei, B.; et al. Upregulation of GSK3β Contributes to Brain Disorders in elderly REGγ-Knockout Mice. Neuropsychopharmacology 2016, 41, 1340–1349. https://doi.org/10.1038/npp.2015.285.
- Murata, S.; Kawahara, H.; Tohma, S.; Yamamoto, K.; Kasahara, M.; Nabeshima, Y.; Tanaka, K.; Chiba, T. Growth Retardation in Mice Lacking the Proteasome Activator. J. Biol. Chem. 1999, 274, 38211–38215. https://doi.org/10.1074/jbc.274.53.38211.
- Barton, L.F.; Runnels, H.A.; Schell, T.D.; Cho, Y.; Gibbons, R.; Tevethia, S.S.; Deepe Jr, G.S.; Monaco, J.J. Immune Defects in 28-KDa Proteasome Activator Gamma-Deficient. J. Immunol. 2004, 172, 3948–3954. https://doi.org/10.4049/jimmunol.172.6.3948.
- Masson, P.; Lundgren, J.; Young, P. Drosophila Proteasome Regulator REGgamma: Transcriptional by DNA Replication-Related Factor DREF and evidence for a Role in Cell Cycle Progression. J. Mol. Biol. 2003, 327, 1001–1012. https://doi.org/10.1016/s0022-2836(03)00188-8.
- He, J.; Cui, L.; Zeng, Y.; Wang, G.; Zhou, P.; Yang, Y.; Ji, L.; Zhao, Y.; Chen, J.; Wang, Z.; et al. REGγ Is Associated with Multiple Oncogenic Pathways in human Cancers. BMC Cancer 2012, 12, 75. https://doi.org/10.1186/1471-2407-12-75.
- He, X.; Li, Y.; Chen, Q.; Zheng, L.; Lou, J.; Lin, C.; Gong, J.; Zhu, Y.; Wu, Y. O-GlcNAcylation and Stablization of SIRT7 Promote Pancreatic Progression by Blocking the SIRT7-REGγ. Cell Death Differ. 2022. https://doi.org/10.1038/s41418-022-00984-3.
- Chen, H.; Gao, X.; Sun, Z.; Wang, Q.; Zuo, D.; Pan, L.; Li, K.; Chen, J.; Chen, G.; Hu, K.; et al. REGγ Accelerates Melanoma Formation by Regulating/β-Catenin Signalling Pathway. Exp. Dermatol. 2017, 26, 1118–1124. https://doi.org/10.1111/exd.13394.
- Li, L.; Dang, Y.; Zhang, J.; Yan, W.; Zhai, W.; Chen, H.; Li, K.; Tong, L.; Gao Xiao and Amjad, A.; Ji, L.; et al. REGγ Is Critical for Skin Carcinogenesis by modulating the Wnt/β-Catenin Pathway. Nat. Commun. 2015, 6, 6875. https://doi.org/10.1038/ncomms7875.
- Zhu, X.; Yang, M.; Lin, Z.; Mael, S.K.; Li, Y.; Zhang, L.; Kong, Y.; Zhang, Y.; Ren, Y.; Li, J.; et al. REGγ Drives Lgr5+ Stem Cells to Potentiate Radiation Intestinal Regeneration. Sci. China Life Sci. 2021. https://doi.org/10.1007/s11427-021-2018-7.
- Rigoulet, M.; Bouchez, C.L.; Paumard, P.; Ransac, S.; Cuvellier, S.; Duvezin-Caubet, S.; Mazat, J.P.; Devin, A. Cell Energy Metabolism: An Update. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148276. https://doi.org/10.1016/j.bbabio.2020.148276.
- Yao, L.; Xuan, Y.; Zhang, H.; Yang Bo and Ma, X.; Wang, T.; Meng, T.; Sun, W.; Wei, H.; Ma, X.; Moses, R.; et al. Reciprocal REGγ-MTORC1 Regulation Promotes Glycolytic in Hepatocellular Carcinoma. Oncogene 2021, 40, 677–692. https://doi.org/10.1038/s41388-020-01558-8.
- Huang, L.; Haratake, K.; Miyahara, H.; Chiba, T. Proteasome Activators, PA28γ and PA200, Play Roles in Male Fertility. Sci. Rep. 2016, 6, 23171. https://doi.org/10.1038/srep23171.
- Clotaire, D.Z.J.; Wei, Y.; Yu, X.; Ousman, T.; Hua, J. Functions of Promyelocytic Leukaemia Zinc Finger (Plzf) in Male Stem Cell Development and Differentiation. Reprod. Fertil. Dev. 2019, 31, 1315. https://doi.org/10.1071/RD18252.
- Gao, X.; Chen, H.; Liu, J.; Shen, S.; Wang, Q.; Clement, T.M.; Deskin, B.J.; Chen Caiyu and Zhao, D.; Wang, L.; Guo, L.; et al. The REGγ-Proteasome Regulates Spermatogenesis partially by P53-PLZF Signaling. Stem Cell Rep. 2019, 13, 559–571. https://doi.org/10.1016/j.stemcr.2019.07.010.
- Magni, M.; Ruscica, V.; Buscemi, G.; Kim, J.-E.; Nachimuthu, B.T.; Fontanella, E.; Delia, D.; Zannini, L. Chk2 and REGγ-Dependent DBC1 Regulation in DNA Induced Apoptosis. Nucleic Acids Res. 2014, 42, 13150–13160. https://doi.org/10.1093/nar/gku1065.
- Shi, Y.; Luo, X.; Li, P.; Tan, J.; Wang, X.; Xiang, T.; Ren, G. MiR-7-5p Suppresses Cell Proliferation and Induces Apoptosis of breast Cancer Cells Mainly by Targeting REGγ. Cancer Lett. 2015, 358, 27–36. https://doi.org/10.1016/j.canlet.2014.12.014.
- Shu, Y.; Long, J.; Guo, W.; Ye, W. MicroRNA 195 5p Inhibitor Prevents the Development of osteoarthritis by Targeting REGγ. Mol. Med. Rep. 2019, 19, 4561–4568. https://doi.org/10.3892/mmr.2019.10124.
- Liu, J.; Yu, G.; Zhao, Y.; Zhao, D.; Wang, Y.; Wang, L.; Liu, J.; Li, L.; Zeng Yu and Dang, Y.; Wang, C.; et al. REGgamma Modulates P53 Activity by Regulating Its Cellular. J. Cell Sci. 2010, 123, 4076–4084. https://doi.org/10.1242/jcs.067405.
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does P53 Induce Apoptosis and How Does This Relate To-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. https://doi.org/10.1038/cdd.2017.169.
- Moncsek, A.; Gruner, M.; Meyer, H.; Lehmann, A.; Kloetzel, P.-M.; Stohwasser, R. Evidence for Anti-Apoptotic Roles of Proteasome Activator via Inhibiting Caspase Activity. Apoptosis 2015, 20, 1211–1228. https://doi.org/10.1007/s10495-015-1149-6.
- Uriarte, M.; sen Nkwe, N.; Tremblay, R.; Ahmed, O.; Messmer, C.; Mashtalir Nazar and Barbour, H.; Masclef, L.; Voide, M.; Viallard, C.; Daou, S.; et al. Starvation-Induced Proteasome Assemblies in the Nucleus Link Acid Supply to Apoptosis. Nat. Commun. 2021, 12, 6984. https://doi.org/10.1038/s41467-021-27306-4.
- Naumova, O.Y.; Palejev, D.; Vlasova, N.V.; Lee, M.; Rychkov, S.Y.; Babich, O.N.; M. Vaccarino, F.; Grigorenko, E.L. Age-Related Changes of Gene Expression in the Neocortex: Preliminary Data on RNA-Seq of the Transcriptome in Three Distinct Cortical Areas. Dev. Psychopathol. 2012, 24, 1427–1442. https://doi.org/10.1017/S0954579412000818.
- Satoh, J.-I.; Yamamoto, Y.; Asahina, N.; Kitano, S.; Kino, Y. RNA-Seq Data Mining: Downregulation of NeuroD6 Serves as a possible Biomarker for Alzheimer’s Disease Brains. Dis. Markers 2014, 2014, 123165. https://doi.org/10.1155/2014/123165.
- Seo, H.; Sonntag, K.-C.; Kim, W.; Cattaneo, E.; Isacson, O. Proteasome Activator Enhances Survival of Huntington’s Disease Model Cells. PLoS One 2007, 2, e238. https://doi.org/10.1371/journal.pone.0000238.
- Jeon, J.; Kim, W.; Jang, J.; Isacson, O.; Seo, H. Gene Therapy by Proteasome Activator, PA28γ, Improves Coordination and Proteasome Function in Huntington’s YAC128 Mice. Neuroscience 2016, 324, 20–28. https://doi.org/10.1016/j.neuroscience.2016.02.054.
- Levy-Barda, A.; Lerenthal, Y.; Davis, A.J.; Chung, Y.M.; Essers, J.; Shao, Z.; van Vliet, N.; Chen, D.J.; Hu, M.C.-T.; Kanaar, R.; et al. Involvement of the Nuclear Proteasome Activator PA28γ in the Cellular Response to DNA Double-Strand Breaks. Cell Cycle 2011, 10, 4300–4310. https://doi.org/10.4161/cc.10.24.18642.
- Liedtke, V.; Schröder, C.; Roggenbuck, D.; Weiss, R.; Stohwasser, R.; Schierack Peter and Rödiger, S.; Schenk, L. LEDGF/P75 Is Required for an Efficient DNA Damage Response. Int. J. Mol. Sci. 2021, 22, 5866. https://doi.org/10.3390/ijms22115866.
- Ali, A.; Wang, Z.; Fu, J.; Ji, L.; Liu, J.; Li, L.; Wang, H.; Chen, J.; Caulin, C.; Myers, J.N.; et al. Differential Regulation of the REGγ-Proteasome pathway by P53/TGF-β Signalling and Mutant P53 in Cancer Cells. Nat. Commun. 2013, 4, 2667. https://doi.org/10.1038/ncomms3667.
- Wang, H.; Bao, W.; Jiang, F.; Che, Q.; Chen, Z.; Wang, F.; Tong, H.; Dai Chenyun and He, X.; Liao, Y.; Liu, B.; et al. Mutant P53 (P53-R248Q) Functions as an Oncogene in Promoting Cancer by up-Regulating REGγ. Cancer Lett. 2015, 360, 269–279. https://doi.org/10.1016/j.canlet.2015.02.028.
- Yang, W.; Yang, X.; Wang, X.; Gu, J.; Zhou, D.; Wang, Y.; Yin, B.; Guo J.; Zhou, M. Silencing CDR1as Enhances the Sensitivity of Breast Cancer to Drug Resistance by Acting as a MiR-7 Sponge to down-Regulate REGγ. J. Cell. Mol. Med. 2019, 23, 4921–4932. https://doi.org/10.1111/jcmm.14305.
- Li, C.; Li, M.; Xue, Y. Downregulation of CircRNA CDR1as Specifically Triggered-Dose Diosbulbin-B Induced Gastric Cancer Cell Death by regulating MiR-7-5p/REGγ Axis. Biomed. Pharmacother. 2019, 120, 109462. https://doi.org/10.1016/j.biopha.2019.109462.
- Gao, X.; Wang, Q.; Wang, Y.; Liu, J.; Liu, S.; Liu, J.; Zhou, X.; Zhou, L.; Chen, H.; Pan, L.; et al. The REGγ Inhibitor NIP30 Increases Sensitivity to chemotherapy in P53-Deficient Tumor Cells. Nat. Commun. 2020, 11, 3904. https://doi.org/10.1038/s41467-020-17667-7.
- Yu, Z.; Wei, X.; Liu, L.; Sun, H.; Fang, T.; Wang, L.; Li, Y.; Sui, W.; Wang, K.; He, Y.; et al. Indirubin-3′-Monoxime Acts as Proteasome Inhibitor: Therapeutic in Multiple Myeloma. EBioMedicine 2022, 78, 103950. https://doi.org/10.1016/j.ebiom.2022.103950.
- Wu, Y.; Wang, L.; Zhou, P.; Wang, G.; Zeng, Y.; Wang, Y.; Liu, J.; Zhang, B.; Liu, S.; Luo, H.; et al. Regulation of REGγ Cellular Distribution and function by SUMO Modification. Cell Res. 2011, 21, 807–816. https://doi.org/10.1038/cr.2011.57.
- Zhang, Y.; Liu, S.; Zuo, Q.; Wu, L.; Ji, L.; Zhai, W.; Xiao, J.; Chen, J.; Li, X. Oxidative Challenge Enhances REGγ-Proteasome-Dependent Degradation. Free Radic. Biol. Med. 2015, 82, 42–49. https://doi.org/10.1016/j.freeradbiomed.2015.01.024.