Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
|
Biochemistry & Molecular Biology
EML4-ALK is an oncogenic fusion protein that accounts for approximately 5% of non-small cell lung cancer cases. Targeted inhibitors of ALK are now the standard of care treatment often leading to an excellent initial response and increased overall survival. However, a variety of resistance mechanisms have been identified that eventually lead to relapse highlighting the need for additional treatment options for these patients.
- EML4-ALK
- NSCLC
- ALK inhibitors
- TKIs
- chemotherapy
1. Epidemiology and Genetics of Lung Cancer
Lung cancer is the leading type of cancer in males and among the top three for females around the globe. Regardless of gender, it is also the leading cause of cancer-related mortality worldwide [1]. Lung cancer is classified as either small cell lung cancer (~15%) (SCLC) or non-small cell lung cancer (~85%) (NSCLC), with the latter further categorized into adenocarcinomas, squamous cell or large cell carcinomas [2]. Lung cancer is strongly associated with tobacco smoking and the introduction of tobacco control programmes has resulted in a sharp decline in both incidence and mortality [3,4,5]. Despite such programmes, smoking continues to be a key risk factor, along with other environmental risk factors (e.g., ionizing radiation, air pollution), and age and genetic risk factors. Notably, less than 10% of lung cancer patients survive for 10 years or more [6,7].
Several genetic changes have been identified as the main drivers of NSCLC, with EGFR and KRAS mutations being ranked as the most common followed by ALK rearrangements [8]. The discovery of oncogenic driver mutations resulted in the development of targeted therapies that have revolutionised the treatment paradigm for NSCLC, resulting in better long-term patient outcomes [9]. Interestingly, NSCLC patients with either an EGFR mutation or ALK rearrangement are more likely to be non- or light-smokers, suggesting that the proportion of these will increase as smoking-related changes reduce [10]. A recent review by the National Institute for Health, identified three molecular subtypes of cancer in non-smokers: piano, a slow growing subtype associated with progenitor cells with few mutations, mezzo-forte, harbouring EGFR-related mutations which grew quicker than the piano group, and forte, which had genomic changes similar to those who smoked, demonstrating that there is heterogeneity in the pathogenesis of tumours in this population of patients. An increased focus on never smoker NSCLC patients will hopefully in time lead to further personalised therapy options for these individuals, who typically present later compared to their smoking counterparts often due to the vague nature of their symptoms [11].
2. ALK Rearrangements
The anaplastic lymphoma kinase (ALK) gene was initially discovered in 1994 in non-Hodgkin’s lymphoma, as a result of a chromosomal translocation involving the nucleophosmin (NPM) gene that led to the formation of the NPM-ALK oncogenic fusion protein [12]. ALK is situated on the short arm of chromosome 2 and encodes a transmembrane receptor tyrosine kinase (RTK). It is highly expressed in neuronal cells during the early stages of cellular development, but expression is almost completely absent in mature cells [13]. Since 1994, ALK rearrangements involving a variety of genes have been reported in several types of malignancies, including colorectal, breast and NSCLC [14,15]. Most prominently, in-frame fusion of ALK with the EML4 gene, which encodes the echinoderm microtubule (MT)-associated protein (EMAP)-like 4 (EML) protein to create the EML4-ALK fusion protein, is found in approximately 5% of NSCLC cases [10].
3. The EML4-ALK Oncogenic Fusion
The EML4 gene is the most common fusion partner of ALK and EML4-ALK was first identified in 2007 in ~7% of Japanese NSCLC patients [16,17]. The EML4-ALK fusion is now reported among ~4–6% of NSCLC patients and has allowed for the development of personalized therapies for ALK+ NSCLC patients [18,19]. A number of diagnostic approaches are currently in use for identification of the EML4-ALK fusion in ALK+ patients. Practice regarding molecular diagnostics will vary widely driven by different health care systems and availability of testing and treatment options resulting in heterogeneity of approaches. In addition, biopsy sample size may be limited and must be handled with care, ensuring tissue is used sparingly. Guidelines vary globally, but the cornerstone of diagnosis is based on fluorescence in situ hybridization (FISH), immunohistochemistry (IHC) or more recently next-generation sequencing (NGS). The United States Food and Drug Administration (FDA) has a list of approved testing techniques as a companion diagnostic for each ALK inhibitor therapy. Testing platforms currently include IHC, FISH and NGS [20]. In Europe, historically the standard of care for detection of an ALK rearrangement is FISH, though in some countries IHC can also be used in conjunction with another approach for confirmation [21,22,23,24,25].
The EML4-ALK oncogenic fusion results from a paracentric inversion on the short arm of chromosome 2 that joins the regions coding for the N-terminus of EML4 to that encoding the C-terminal kinase domain of ALK [16]. Different breakpoints in the EML4 gene give rise to distinct EML4-ALK variants (Figure 1) [16,26]. To date, more than 15 variants have been discovered in NSCLC, some of which have multiple isoforms as a result of alternative splicing. However, variants 1 (V1) and 3 (V3) are the most common by some margin, and together represent approximately 80% of EML4-ALK cases [16,26,27]. Routinely variant testing is not assessed clinically in most healthcare systems globally.
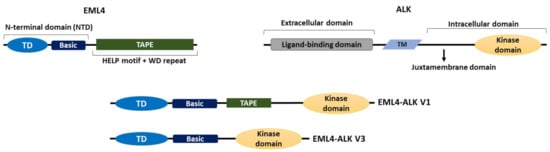
Figure 1. The domain organizations of EML4, ALK and EML4-ALK V1 and V3. A cartoon illustrating the domain organizations of EML4, ALK and the two most commonly occurring EML4-ALK variants, the long V1 (E13; A20) and the short V3 (E6; A20); “E” and “A” represent the exons of EML4 and ALK, respectively, that are fused in each variant. Both variants contain the intracellular kinase domain of ALK and the TD and basic region of EML4. V1 also expresses a truncated TAPE domain from EML4 whereas V3 completely lacks the TAPE domain. TM = transmembrane domain, TD = trimerization domain.
There are six human EML proteins, named EML1 to EML6. These are homologues of the echinoderm microtubule-associated protein (EMAP), which was isolated from unfertilised sea urchin eggs in 1993 [28]. EML1 to EML4 have a similar organization with an N-terminal domain (NTD) made up of a coiled-coil followed by a region rich in basic residues. Structural evidence indicates that the coiled-coil leads to trimer assembly, hence this motif has been called a trimerization domain (TD). Localization studies demonstrate that the TD together with the basic region is essential for microtubule binding of these EMLs [29,30]. Meanwhile, a series of tryptophan-aspartate (WD) repeats fold into two seven-bladed β-propellers that, along with a hydrophobic EML-like protein (HELP) motif, form the C-terminal tandem atypical propeller in EMLs (TAPE) domain of EML1 to EML4 [30,31]. EML5 and EML6 are somewhat different in organization, lacking the NTD and having three contiguous repeats of the TAPE domain encoded in a single polypeptide [31]. Therefore, consistent with the importance of the NTD for microtubule binding, the lack of NTD in EML5 and EML6 suggests that they may be unable to bind microtubules, but this remains to be tested.
The ALK protein comprises an extracellular domain required for ligand binding, a transmembrane sequence, and an intracellular domain, which contains the tyrosine kinase [32]. All EML4-ALK variants contain the complete ALK tyrosine kinase domain but lack the transmembrane and extracellular regions. Importantly though, they differ in the amount of the EML4 protein they include, due to distinct breakpoints in the EML4 gene. For example, V1, V2 and V4 possess the EML4 NTD and part of the TAPE domain and are known as the long variants, while V3 and V5 have some or all of the NTD, but none of the TAPE domain and are referred to as the short variants [30] (Figure 1). The presence of the TAPE domain in the long EML4-ALK variants significantly reduces the stability of the fusion proteins, and deletion of different parts of the TAPE domain from the long variants resulted in a stability profile similar to that of the short variant, V3, in which the TAPE domain is completely absent [33].
Considerable research has focused on elucidating the mechanisms behind EML4-ALK fusion-driven cancer progression, largely due to the fact that they are the most common ALK rearrangement in NSCLC. Firstly, given that all variants contain the EML4 TD, it is widely assumed that trimerization of EML4-ALK monomers via the TD facilitates the trans-autophosphorylation of ALK, which gives rise to a constitutively active tyrosine kinase [16,29,34]. However, in other respects there are several important differences in the biological properties of the long and short variants.
EML4-ALK V1 and V3 are the two most common variants, and are representative of the long and short variants, respectively. Although both variants contain the TD and basic region of EML4, only V3 localizes to microtubules in a similar manner to the wild-type EML4 protein, while V1 is strictly cytoplasmic, potentially because the incomplete TAPE domain sequence perturbs binding to microtubules [29]. As previously mentioned, the presence of the partial TAPE domain within the long EML4-ALK variants reduces their stability and they require the heat-shock protein 90 (HSP90) chaperone to maintain their expression. Hence, it is also possible that association with HSP90 perturbs interaction of the long variants with microtubules [35]. However, recent evidence suggests that the presence of the 12N blade region within the TAPE domain in the long variants inhibits their microtubule localization, because deletion of this region results in V1 associating with microtubules [36]. These differences in localization and stability could explain the profound variability in both prognosis and response to targeted treatment for patients with different variants. For example, cells expressing the short variant V3, exhibit a much lower sensitivity to the ALK tyrosine kinase inhibitor (TKI), crizotinib, than those expressing the long variant V2 [33,35]. Moreover, Christopoulos et al. (2018) found that patients with V3 exhibited enhanced metastasis and reduced overall survival (OS) compared to those with V1 or V2, supporting the hypothesis that EML4-ALK V3, and potentially short variants more broadly, are more aggressive variants, resulting in poorer patient outcomes [37].
Intriguingly, the oligomerization and activation of both EML4-ALK V1 and V3 enables the formation of cytoplasmic compartments, described as granules or foci, that contain downstream components of several ALK-dependent signalling pathways, including those of the RAS/MAPK and JAK/STAT pathways, along with the EML4-ALK protein [36,38,39]. Both ceritinib and lorlatinib, two ALK inhibitors, can dissolve these cytoplasmic foci and redirect V3, but not V1, to microtubules, which mirrors the localization of a catalytically inactive V3 mutant. However, this does not rule out the possibility that active V3 also localizes to microtubules as well as within cytoplasmic foci. Nonetheless, the formation of these cytoplasmic granules appears to be dependent on ALK activity. Furthermore, constitutively active ALK mutants stabilized these foci even in the presence of ALK inhibitors [36]. Interestingly, the disruption of the EML4 portion of the fusion protein prevents the formation of cytoplasmic granules, highlighting the potential importance of EML4 in forming these condensates [39].
4. Targeted ALK Inhibitors and Resistance Mechanisms
Targeted ALK inhibitors have become the standard of care treatment for ALK positive (ALK+) NSCLC patients [40,41]. A growing number of ALK TKIs have been approved for use, including first- (crizotinib; [18,19,42,43]), second- [alectinib, ceritinib, brigatinib; [44,45,46,47,48] and third-generation (lorlatinib, ensartinib, entrectinib; [49]) inhibitors with each consecutive generation having enhanced clinical properties such as blood-brain-barrier penetration. However, despite their initial efficacy, patients often become resistant to ALK TKIs. It is thus imperative to understand the mechanisms behind this resistance and develop alternative approaches to treatment [19,43,50]. Furthermore, fourth-generation inhibitors such as TPX-0131 are also in pre-clinical studies, and preliminary data has indicated that TPX-0131 has high potency against both wild-type ALK and ALK that is resistant to previous generation ALK inhibitors [51].
To date, several different ALK mutations have been identified that promote resistance to ALK inhibitors, with the majority sitting within the ATP-binding pocket of the ALK catalytic domain. Interestingly, these can confer differential sensitivity to distinct ALK TKIs [52]. For instance, the ALK F1174V missense mutation confers resistance to ceritinib but sensitivity to alectinib, whereas conversely the ALK I1171S mutation leads to resistance to alectinib but sensitivity to ceritinib [53,54]. Meanwhile, an ALK F1245C mutation promotes resistance to the first-generation ALK inhibitor, crizotinib, while G1202R is the most common mutation observed in tumours resistant to second generation ALK TKIs [52,55]. Hence, characterization of the ALK sequence following development of resistance to one ALK TKI would facilitate selection of the most suitable subsequent treatment.
Other than ALK catalytic site mutations, additional resistance mechanisms have been described. For example, ALK amplification is also able to promote crizotinib resistance [56,57]. Furthermore, tumours can switch to ALK-independent growth through activation of “bypass” signalling pathways for growth and survival. Some of the most common bypass pathways involve EGFR, MAPK or IGF-IR [57,58,59]. Finally, the co-occurrence of EML4-ALK with other genetic changes—for example, TP53 mutation—can serve as a resistance mechanism by promoting cell survival and other tumour-related adaptations such as upregulation of MYC. Indeed, overexpression of MYC, a transcriptional regulator of multiple cancer-related processes, has been linked to resistance to crizotinib and alectinib [60,61].
This entry is adapted from the peer-reviewed paper 10.3390/cancers14143452
This entry is offline, you can click here to edit this entry!