Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Diet and lifestyle factors greatly affect health and susceptibility to diseases, including cancer. Stem cells’ functions, including their ability to divide asymmetrically, set the rules for tissue homeostasis, contribute to health maintenance, and represent the entry point of cancer occurrence. Stem cell properties result from the complex integration of intrinsic, extrinsic, and systemic factors. In this context, diet-induced metabolic changes can have a profound impact on stem cell fate determination, lineage specification and differentiation.
- diet
- stem cells
- cancer stem cells
- caloric restriction
- asymmetric division
- nutrients
- autophagy
- mTOR
- SIRT1
1. Introduction
Lifestyle plays a crucial role in health and cancer development. From before conception and during pregnancy, infancy and early childhood, the environment programs an individual’s susceptibility to developing diseases, including tumors, and continues to exert its influence throughout the entire lifespan. Studies on migrant populations support the crucial role of the environment in cancer susceptibility. Cancer rates in migrant populations start shifting from those of the country of origin already after the first decade of migration and will resemble those of the host country within one to two generations [1], highlighting the relevance of changes in dietary and other lifestyle factors in cancer initiation and progression. Forty years have passed since Doll and Peto, with their landmark paper, acknowledged that cancer is a largely avoidable disease and identified smoking and diet as important determinants of health and disease [2]. Two thirds of all cancers can be prevented by refraining from smoking, eating a healthy diet, avoiding excessive weight gain and being physically active [3]. Yet, as studies of molecular pathological epidemiology have highlighted, the exogenous lifestyle factors interact with the individual molecular arrangement, where each neoplasm is the result of a dynamic interplay among environment, host and tumor [4].
Dietary patterns, foods and bioactive food compounds have been found to significantly modify lifespan and health in diverse organisms such as yeast, worms, flies, and mammals and to affect cancer risk and tumor growth, directly or indirectly participating in the cancer process with a profound impact on all cancer’s hallmarks [5][6][7][8][9]. Food quality (what we eat), food quantity (how much we eat) and meal frequency (when we eat) are all aspects that weigh in on the impact of diet on our health system.
Several lines of evidence converge on the idea that stem cells (SCs) are among the major players in orchestrating the response of our body to nutrients, mainly due to their key role in tissue homeostasis. SCs are undifferentiated cells with the potential to generate, upon cell division, different cell types in an organism. They are characterized by unique metabolic features compared to differentiated cells [10][11][12], and they can potentially maintain their undifferentiated state all their life while generating offspring cells committed to differentiation in response to specific needs for tissue homeostasis. In doing so, tissue SCs not only utilize nutrients for their metabolic needs but also adapt their functions, such as self-renewal, autophagy, or differentiation, to the metabolic environment and nutrient availability [12][13][14]. On the other hand, their relatively long lifespan, whereas indispensable to fulfill their function in tissue turnover, holds the back of the coin of continuously being exposed to environmental factors, including diet, and progressively accumulating cell damage at the genetic and epigenetic level, with significant consequences on gene and protein expression and molecular pathways [15][16]. The consequent perturbation in the balance between maintenance and loss of SCs characteristics can be further propagated in the offspring cells, thus inducing alterations in their functionality. In addition, the molecular pathways in charge of sensing nutrient availability also control key SC functions, such as protein synthesis, self-renewal, autophagy, and differentiation, mediating the effects of harmful dietary factors and possibly pushing SCs toward the downhill of cancer transformation, thus generating cancer stem cells (CSCs) [17]. Similarly, some nutrients and dietetic regimens have been studied for their beneficial effects on the regulation of SCs and tissue homeostasis that may result in the inhibition of the cancer transformation process and progression and may prevent the onset of drug resistance [18][19].
Nutrients are normally crucial in SC physiology due to the ability of many nutrient-derived metabolites, released during the catabolic process, to induce chromatin reshaping, epigenetic modifications and gene expression modulation [20]. In this context, for example, glucose has been reported to act as an SC fate regulator, controlling key phases of embryo development [20][21][22]. Similarly, glycolysis and acetyl-CoA have been described to play an active role in the maintenance of pluripotency through induction of histone acetylation; accordingly, modifications of glycolysis or administration of acetyl-CoA precursors can inhibit differentiation and histone deacetylation [11]. Nutrients also serve as donors for moieties involved in post-translational modifications. One example is represented by the hexosamine biosynthetic pathway (HBP) that generates uridine diphosphate GlcNAc (UDP-GlcNAc) for the O-GlcNAcylation of serine and threonine residues embedded in cytoplasmic, nuclear and mitochondrial proteins. In SCs, these post-translational modifications link glucose and nutrient availability with the epigenetic regulation of cell fate determination and differentiation [23]; alterations in this finely regulated process are at the basis of cancer transformation as well as neurodegeneration [24].
Amino acids (AAs) are also involved in SC self-renewal, maintenance of pluripotency and differentiation ability [25]. Several essential AAs (EAAs) are required for the maintenance of both embryonic [26] and adult SCs [27] and their abundance has been demonstrated to increase proliferation, without affecting stemness [21]. In Embryonic Stem Cells (ESCs), threonine and methionine participate in the maintenance of the epigenetic regulation of pluripotency as methyl-group donors for histone methylation. In the absence of methionine or threonine, intracellular S-adenosyl methionine (SAM) levels decrease, DNA and histones methylation is reduced, thus activating p53 expression and inhibiting pluripotency markers and differentiation [25]. Among EAAs, branched-chain amino acids (BCAAs), leucine, isoleucine and valine, represent a major source of acetic acid, hence playing a central role in histone acetylation, epigenetic modifications, transcriptional regulation, and autophagy [5]. BCAAs have been described to have multiple roles in SC physiology and health. Indeed, they have been found to improve satellite cell function [28], Neural Stem Cell (NSC) differentiation and antioxidant defense [29], as well as proliferation and survival of hematopoietic stem cells (HSCs) [25]. Similarly, non-essential AAs (NEAAs) have been shown to be required for skeletal muscle stem and progenitor cell function [30]. Proline, which is abundant in meat and fish, has been shown to preserve the identity of ES cells and to be necessary for their self-renewal and differentiation [31]. Increased proline availability induces mouse ES cells to acquire mesenchymal-like features, such as increased motility and invasiveness (“embryonic-stem-to-mesenchymal like transition”-esMT), and to promote the synthesis of proline-rich proteins, such as collagen.
Fatty acids (FAs) represent another class of nutrient-derived molecules; their importance for SC physiology is demonstrated by the presence of a specific lipidome signature found in certain adult SCs, playing a primary role in the regulation of processes such as quiescence and self-renewal, symmetric-asymmetric division, differentiation, cell-niche interaction and cell fate determination [32]. In SCs, a well-balanced combination of FA synthesis and FA oxidation (FAO) is indispensable, and inhibition of one or the other leads to SC exhaustion [32]. In particular, FAO was shown to be necessary for the maintenance of quiescence of HSCs, NSCs, Intestinal Stem Cells (ISCs) and skeletal Muscle Stem Cells (MuSCs). Studies of in vitro lipid supplementation or deprivation have demonstrated their effects on SC proliferation and/or differentiation potential [33][34][35], bioenergetics [36] and pluripotency [37].
2. Molecular and Cellular Effects of Diet on Stem Cells
The extraordinary potential of SCs, either embryonic or adult, resides in their ability to provide tissues with brand new cells throughout life, given their capacity to divide symmetrically or asymmetrically and lead either to SC self-renewal or differentiation. While the underlying balance of this process is controlled by endogenous mechanisms of development and gene regulation, exogenous signals from the microenvironment, including nutrients, can significantly affect SC specification, differentiation and performance, thus acting on aging and disease. Indeed, nutrients may act directly on SCs or indirectly by regulating the SC niche (non-autonomously). Moreover, nutrients can regulate hormone production, which in turn can influence the behavior of SCs and their niche. In response to these direct and indirect stimuli, SCs activate signaling pathways, reprogram their metabolism and gene expression, converting the dietary input into fate decisions (Figure 1).
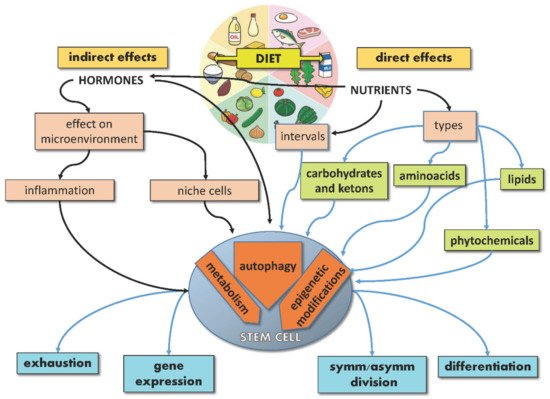
Figure 1. Schematic illustration showing direct and indirect effects of diet on stem cells.
SC features regulated by nutrients include symmetric/asymmetric division balance, genome and epigenome integrity, gene expression, metabolism and oxidative status, autophagy, self-renewal, differentiation, and exhaustion. SCs adapt their proliferation to nutrients and growth factors’ availability to undergo cell division when nutrients are sufficient. Mechanistically, this tight balance depends on “master regulators”, such as mTORC1, which can sense nutrients and regulate both metabolism and SC fate [14]. On the other hand, intracellular metabolites, such as acetyl-CoA, regulate both metabolic pathways and epigenetic processes [38], thus connecting diet and metabolism with SC functions. This connection is particularly relevant in fate determination for different types of SCs, as SC self-renewal can be achieved by modifying calories or nutrients [16] and skeletal MuSCs from calorie-restricted mice are more efficient in inducing muscle regeneration than those from ad-libitum-fed mice in transplantation experiments [12]. Similarly, induced pluripotent stem cells (iPSCs) can be reprogrammed by manipulating metabolic pathways [14]; whereas MuSC activation requires the shift from fatty acid oxidation (FAO) to glycolysis [39], the upregulation of glycolytic pathways, as well as the inhibition of mitochondrial activity, may be sufficient to induce stemness features in iPSCs and ESCs [40][41][42]. In vivo, extracellular glucose was found to be capable of modifying gene expression in mouse trophoblast SCs, inducing gene expression changes both before and after differentiation [43]. Notably, crucial stemness master regulators, such as OCT4 and OCT1, have been reported to regulate the expression of glycolytic genes [44][45], thereby strengthening the interdependence between metabolism and stemness maintenance. Intriguingly, even CSC characteristics can be modified by diet, as caloric restriction (CR) has been found to inhibit their cancerogenic and metastatic potential [9].
For both SCs and CSCs, the main molecular node connecting diet and function is represented by the AMPK-mTOR-SIRT1 pathway. When the AMP-activated protein kinase (AMPK) senses low cellular ATP levels, induced by fasting (or exercise), it is phosphorylated by the serine–threonine kinase liver kinase B1 (LKB1) and, in turn, it directly or indirectly modulates enzymes involved in glucose [46] and lipid metabolism [47], as well as the mTOR pathway, thus regulating proteostasis and cell growth [48]. AMPK also targets proteins controlling apoptosis (through direct phosphorylation of p53), cell proliferation (cyclin D1), cell polarity [48], differentiation [49], response to hypoxia (HIF1α) and autophagy [50], hence affecting SC fate [48][51]. Moreover, AMPK increases cellular NAD+, which activates the NAD-dependent histone deacetylase SIRT1, affecting gene expression [52], protein synthesis and SC self-renewal [53]. These molecular events ultimately impact SC “performance” and prevent SC transformation into CSCs. Accordingly, mTOR, AMPK and SIRT1 have been found deregulated in tumors [48][54][55] and in correlation with lifestyle factors lowering ATP, such as CR and exercise, which activate AMPK and are associated with lower cancer risk [6].
2.1. Autophagy and Liquid-Liquid Phase Separation
Autophagy is a conserved homeostatic lysosome-mediated and highly selective self-degradation process that eliminates misfolded or undesired macromolecules and damaged organelles [56]. Further, in stress conditions, including nutrient deprivation, high temperature and exercise, cells digest and recycle self-components to generate energy and building blocks to foster cell survival. The ability to perform autophagy is inextricably linked with aging and health since a progressive impairment of this function, due to reduced autophagy-related proteins and decreased dispatch to lysosomes, is a common denominator of aging tissues and age-associated diseases [56]. Accordingly, its activation through pharmacological treatments or dietetic regimens (such as CR) increases lifespan and health [7][51].
Autophagy is an indispensable process for SCs since it plays a role in maintaining stemness [57] and SC function [58]. In adult and embryonic SCs, the preservation of cellular functions entails the punctual elimination of damaged or detrimental proteins as well as flawed organelles that accumulate with age or pathological conditions. By removing undesired material from the cytoplasm, autophagy not only copes with stress conditions but also fulfills basal physiological needs, contributing to the maintenance of SC functions such as quiescence, self-renewal, activation, metabolism, and differentiation, hindering cellular decline and senescence [57][58][59][60].
In this process, once macromolecules with specific chemical and physical characteristics reach a threshold concentration, they segregate into the cell in a process known as “liquid-liquid phase separation” (LLPS), by undergoing liquid-gel or gel-solid transition [61]. In particular, gel-like aggregates can trigger the generation of autophagosomal membranes, which precisely recognize, envelope and shuttle them to lysosomes in a multistep process that culminates in the generation of autolysosomes [59][61]. Whereas LLPS plays a role in several steps of the autophagic process, including autophagosome assembly, modulation of TORC1 activity and the sorting of proteins for degradation, stress conditions, including nutritional modifications, affect both LLPS and the autophagic process.
mTOR and PKA are the main inhibitors, whereas AMPK and SIRT1 are the main activators of this process [62]. In particular, mTOR and AMPK, being functionally located at the crossroads between sensing nutrient availability, regulating metabolic pathways, and controlling autophagy, are able to master these functions in a concerted way. mTORC1 is a multiprotein complex in which the mTOR serine/threonine kinase interacts, among others, with Raptor [63]. In the presence of abundant AAs or high cellular ATP levels and growth factors, two different small GTPases, Rags (Ras-related GTP-binding proteins) and Rheb (Ras homolog enriched in the brain), promote mTORC1 translocation from the cytoplasm to the lysosomal surface [63]. Once activated, mTOR phosphorylates several substrates, such as eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) and the ribosomal S6 kinase (S6K1), regulating the synthesis of modulators of cell growth, angiogenesis and tumorigenesis, such as MYC, cyclin D1 and hypoxia-inducible factor 1a (HIF1α) [48]. Concomitantly, activated mTOR phosphorylates the three autophagy modulators, autophagy-related protein 13 (ATG13), Unc-51-like autophagy activating kinase 1 (ULK1) and the Vacuolar protein sorting 3 (VPS34), thus inhibiting their assembly into the autophagy-initiating complexes and preventing autophagosome formation by sequestering TFEB/TFE3 into the cytoplasm [61]. In contrast, nutrient deprivation induces mTOR inactivation and switches cell metabolism towards a catabolic mode, stimulating the activity of the master lysosomal/autophagic transcription factors TFEB and TFE3 [61] and leading to ATG13/ULK1 dephosphorylation/activation and autophagy stimulation (Figure 2).
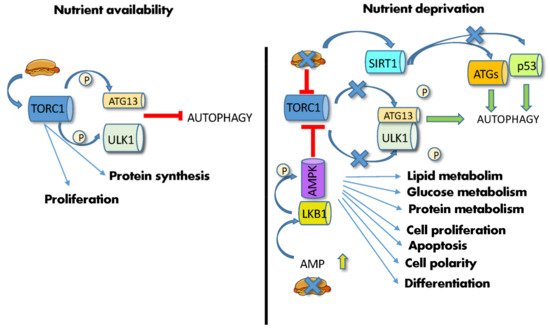
Figure 2. Effects of diet on autophagy. (Left) high nutrient availability activates mTORC1, which phosphorylates ATG13 and ULK1, thus inhibiting autophagy. (Right) nutrient deprivation and low energy supply leads to increase in AMP and AMPK phosphorylation by LKB1, leading to mTOR inhibition, ATG13/ULK1 dephosphorylation/activation and initiation of autophagy. Low ATP levels also activate SIRT1, which induces the deacetylation of the autophagic ATG proteins, and p53. Abbreviations: AMP: adenosine monophosphate; AMPK: 5′ AMP-activated protein kinase; ATG13: autophagy-related gene; LKB1: liver kinase B1; P: phosphate; p53: tumor protein P53; SIRT1: silent information regulator 1; TORC1: mechanistic target of rapamycin complex; ULK1: Unc-51-like autophagy activating kinase 1.
Similarly, low energy supplies and ATP levels induce AMPK phosphorylation via LKB1, leading to mTOR inhibition [48] (Figure 2). Furthermore, low ATP levels increase NAD+ and stimulate a third autophagy player, the NAD+-dependent deacetylase SIRT1. This enzyme mediates the deacetylation of many effectors, including histones, autophagic Atg regulators [7], the p53 tumor suppressor, the DNA repair factor Ku70 and the FOXO3 transcription factor, hence affecting transcription, autophagy, apoptosis, DNA repair, and activation of longevity genes [64]. FOXO3a also mediates autophagy induced by food deprivation in HSCs but not in their more differentiated progeny [65]. Together with mTORC, SIRT1 fosters the expansion of gut adult SCs during CR [53].
Both mTOR and AMPK are crucial to maintaining SC-specific functions. mTOR ensures HSCs quiescence by lowering ROS [66] and stimulates NSC differentiation [67] and myogenesis [68]. In mouse MSCs, mTOR plays a central role in determining cell fate and lineage-specification between adipogenic and osteogenic commitment as follows: activation of mTOR promotes differentiation into adipocytes through the activation of adipocytic genes [69], whereas mTOR inhibition counteracts MSC aging and maintains MSC osteogenic potential [70]. Excessive alcohol consumption leads to osteopenia and reduced osteogenic differentiation of MSCs in mouse models through systematic activation of mTOR, leading to an increase in peroxisome proliferator-activated receptor γ (PPAR-γ) and a reduction of genes responsible for differentiation, such as runt-related transcription factor 2 (RUNX-2) [71]. In epidermal SCs (EpdSCs), WNT-induced hyperproliferation leads to SC exhaustion and aging via mTOR activation [72]. Similarly, in several adult SCs, including MSCs, MuSCs and ISCs, AMPK not only regulates metabolic pathways but also affects differentiation [46][73][74], and its activation reverts mouse epiblast SCs to naive cells [75].
In several types of adult SCs, including MuSCs and HSCs, autophagy is constitutively active and contributes to the maintenance of SC features and the prevention of senescence. Conversely, impairment of autophagy, due to aging or genetic alterations, induces SC senescence caused by failure of proteostasis, compromised mitochondrial function, oxidative stress [57][58], and increased ROS production, leading to SC exhaustion and aging [76]. In particular, autophagy has been shown to play a crucial role in the maintenance and genetic integrity of Lgr5+ ISCs, responsible for intestinal epithelium repair and integrity, either in physiologic or stress-induced conditions. Unlike all intestinal cells, in Lgr5+ ISCs loss of Atg7 leads to increased oxidative stress, altered interaction with the microbiota and impaired DNA repair. As we will better discuss in the following sections, in the mouse intestine, CR inhibits mTORC1 in the niche of Paneth cells, leading to activation of AMPK/SIRT1 and their signaling cascade in the adjacent ISCs [53]. Noticeably, fasting sustains ISC viability and intestine function in the presence of high doses of chemotherapeutic drugs [77]. This is due to the Atg7-dependent ability of Lgr5+ ISCs to repair irradiation-induced DNA damage more effectively than their more differentiated progenitors. Accordingly, fasting-stimulated autophagy prevents doxorubicin and oxaliplatin-induced DNA damage in ISCs, highlighting the role of diet-induced autophagy in SC performance and chemoprotection [78].
In CSCs from several types of tumors, including glioblastoma and colon, mTOR upregulation has been shown to be responsible for self-renewal and tumorigenicity, not only suggesting that sustained stimulation of mTOR induced by hypercaloric diet might trigger SC transformation but also defining mTOR as a potential target for anticancer therapies [79]. However, in CSCs, autophagy plays a pivotal role in enabling plasticity, viability and proliferation, having both promoting and suppressing effects on their activity and survival [80].
Given the role of autophagy in SC performance and health maintenance, a good deal of attention has been focused on finding dietetic strategies or functional foods able to boost this pathway. At the molecular level, AMPK and SIRT1 stimulation of AKT and mTORC1 inhibition are predicted to favor autophagy. From this perspective, CR, fasting, and a ketogenic diet favor SC autophagy by modulating AMPK and mTOR [7][8][51][81][82]. Conversely, a high-fat diet (HFD) inhibits the AMPK pathway in MuSCs, thus compromising cell activation and muscle regeneration [83]. Similarly, low-protein plant-based diets have been found to reduce IGF-1-mediated activation of the AKT-mTORC1 pathway [84]. Some phytochemicals stimulate autophagy, through SIRT1, AMPK or mTOR pathways in both normal and CSCs [7][51][85]. The activation of SIRT1 by thymoquinone, ferulic acid and melatonin has been demonstrated to act on MSCs and preserve bone mass [86]. Spermidine, a polyamine abundant in the Mediterranean diet and found in whole grains, corn, mushrooms, legumes, soy products, and aged cheese, is able to activate autophagy by acting on the same pathways [87]. Resveratrol, found in berries, grapes, pine nuts, and legumes, activates AMPK and SIRT1 [5][7] and inhibits mTOR [88] in different types of SCs.
The ability of some phytochemicals to target CSC autophagy has made them attractive as potential anticancer treatments. Epigallocatechin gallate (ECGC), quercetin and genistein, among all, have been found to hamper AKT function and hence the mTOR pathway [89]. Moreover, ECGC activates AMPK in human breast CSCs, leading to suppression of mTOR, inhibition of growth and up-regulation of the cyclin-dependent kinase inhibitor p21 [90]. Similarly, curcumin was reported to suppress tumorigenic features in CSCs from glioblastoma [91] and liver cancer [92] by acting on mTOR-dependent Atg activation.
2.2. Stem Cell Exhaustion
Several intrinsic factors (DNA damage, altered energy metabolism and mitochondrial function, increased ROS levels caused by spillage of electrons from oxidative phosphorylation, accumulation of misfolded proteins), as well as extrinsic determinants (alterations of the SC niche, modifications of systemic and local factors), contribute to SC functional decline and exhaustion by inducing apoptosis or senescence, which leads to a drop in their self-renewal ability and regenerative potential [93]. In particular, this leads to progenitor cell exhaustion, caused by a decrease in the SC asymmetric division rate, which in turn is responsible for tissue and organismal aging and age-related diseases.
Diet and lifestyle factors can significantly influence both intrinsic and extrinsic factors involved in this process. Dietetic interventions, including CR, were reported to up-regulate metabolic genes, such as glutathione peroxidase, catalase and superoxide dismutase (SOD), and to modulate the expression of genes involved in the mitochondrial function and biogenesis, such as peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC1), endothelial nitric oxide synthase (eNOS), SIRT1 and mitochondrial transcription factor A (TFAM), hence counteracting cellular oxidative stress and regulating mitochondrial activity [94]. In addition, nutritional regimens have been demonstrated to modulate autophagy, displaying a protective function against the accumulation of misfolded proteins and molecular (including DNA) damage [93].
In Drosophila lymph glands, CR and intermittent fasting have been shown to trigger progenitor cell differentiation, including blood cell progenitors, affecting the cellular immune response and counteracting SC exhaustion [95]. Moreover, in the animal world, from worms to mammals, CR slows down aging, likely by reducing ROS and by inhibiting the mTOR pathway. Of note, the mTOR pathway is associated with cell hyperfunction and accelerated senescence, thus leading to an enhancement of SC exhaustion [96]. Fasting also promotes the FAO and improves the function of ISCs during aging [97]. Olive oil consumption has been associated with beneficial effects on virtually all aging-associated processes [98], including SC exhaustion [99]. These benefits have been attributed in part to its high content of monounsaturated FAs and other highly bioactive components, including phenolic compounds such as hydroxytyrosol, tyrosol, caffeic acid, oleuropein aglycone and oleocanthal [99]. Importantly, it has been shown that oleuropein, a polyphenolic compound found in olive oil and olive leaves, stimulates osteoblastogenesis while inhibiting adipogenesis, by enhancing the osteoblastic phenotype instead of the adipocyte differentiation from MSC progenitors in human bone marrow [100]. Therefore, olive oil consumption has been associated with slower skeletal aging, a complex process in which the continuous recruitment of progenitor cells toward adipogenic differentiation leads to their rapid exhaustion and reduced recruitment into the osteoblastic lineage cells and decreased bone formation.
2.3. Epigenome and Gene Expression
In the last two decades, epigenetic modifications have been associated with SC identity, aging and CSC transformation [101]. Further, nutrition has emerged as a fundamental regulator of the epigenome and gene expression, hence affecting cell metabolism and health [102]. This is achieved by the peculiar capacity of some metabolites to either directly associate with chromatin or indirectly modulate chromatin-modifying enzymes. In SCs, epigenetic modifications of DNA and DNA-associated histones orchestrate their function and fate decisions. Therefore, inputs from the diet can lead to altered chromatin structure and gene expression [26][39][103][104] in embryonic and adult SCs, thus affecting processes such as embryonic development, cell fate determination, cell differentiation, immune function, aging, and oncogenic transformation [105][106], making nutrition, metabolism, epigenetics and SC functions closely correlated to each other [107]. Similarly, in CSCs, some metabolic pathways induce specific epigenetic modifications [10], which discriminate metastatic from primary CSCs and regulate cellular plasticity and aggressiveness [108].
Nutrients introduced by diet are processed into simple metabolites through digestion and, once systemically available, can be uptaken by SCs. These biomolecules can be further catabolized by metabolic enzymes into substrates or cofactors utilized by chromatin-modifying enzymes. In some cases, these enzymes can relocate to the nucleus and catalyze chromatin modifications in the presence of specific cofactors [109]. Nutrient-induced epigenetic modifications may affect both histones (acetylation, acylation, ADP-ribosylation glycation, glycosylation, hydroxylation, methylation, phosphorylation, sumoylation and ubiquitylation) and DNA (methylation and glycation), either enzymatically or non-enzymatically. Metabolites can function as co-factors or substrates for enzymes catalyzing either the addition (“writers”) or the removal (“erasers”) of tagging groups [52]. The newly added chromatin “tag” can induce structural chromatin modifications and phase-separation among differently structured/activated chromatin regions, or it can be recognized and bound by effector proteins (“readers”), modulating gene expression and SC fate (Figure 3). Diet can also modify the expression of epigenetic readers, hence affecting normal SC and CSC function [110].
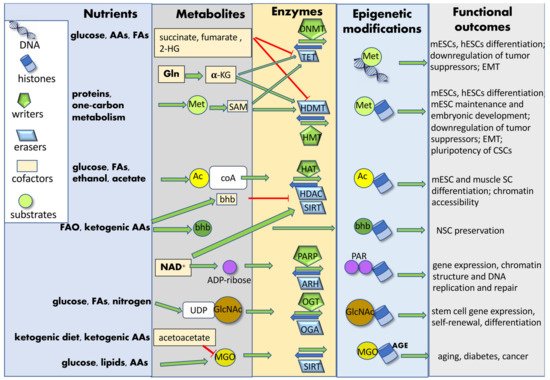
Figure 3. Nutrient-induced epigenetic modifications. Nutrients introduced by diet are processed into simple metabolites and further catabolized by SC metabolic enzymes into substrates or cofactors utilized by chromatin-modifying enzymes. Nutrient-induced epigenetic modifications may affect both histones and DNA, either enzymatically or non-enzymatically. Metabolites can function as co-factors or substrates for enzyme catalyzing either the addition (“writers”) or the removal (“erasers”) of tagging groups. These chromatin modifications finally affect SC gene expression and fate determination. Abbreviations: AAs: amino acids; AGE: advanced glycation end-products; Ac: acetyl group; Ac-CoA: acetyl-coenzyme A; ADP-ribose: adenosine diphosphate ribose; ARH: ADP-ribosyl-hydrolases; bhb: β-hydroxybutyrate; CSCs: cancer stem cells; DNMT: DNA-methyl transferase; EMT: epithelial–mesenchymal transition; FAs: fatty acids; FAO: fatty acid oxidation; GlcNAC: N-acetylglucosamine; Gln: glutamine; HAT: histone acetyltransferase; HDAC: histone deacetylase; HDMT: histone demethylases; hESCs: human embryonic stem cells; 2-HG: 2-hydroxyglutarate; HMT: histone methyltransferase; α-KG: α-ketoglutarate; mESCs: mouse embryonic stem cells; Met: methionine; MGO: methylglyoxal; NAD+: nicotinamide-adenine-dinucleotide; NSC: neural stem cell; OGA: O-linked GlcNAc hydrolase; OGT: O-linked GlcNAc transferase; PAR: poly-ADP-ribose group; PARP: poly-ADP-ribose polymerase; SAM: S-adenosyl methionine; SC: stem cell; SIRT: sirtuin; TET: Tet methyl-cytosine dioxygenase; UDP: uridine diphosphate.
Methylation at DNA and histone levels is the most well-characterized epigenetic modification. Methionine, together with threonine and metabolites from the one-carbon pathway, such as folate, is a fundamental source of intracellular S-adenosylmethionine (SAM), which acts as a donor of methyl groups for DNA and histones [52]. These modifications modulate the expression of pluripotency genes in adult, fetal SCs and CSCs [111] and regulate mouse ESC maintenance and embryonic development [112]. Accordingly, folate and methionine deficiencies have been found associated with reduced histone methylation, gene expression modifications and neural tube closure (NTC) defects [113]. Conversely, reduction in histone and DNA methylation following methionine restriction, which can be achieved by reducing meat and switching to vegetable-enriched diets, has been shown to affect gene expression and to have beneficial effects on health and longevity [114]. In Triple Negative Breast Cancer (TNBC) CSCs, obesity was found to regulate methylation via induction of an epigenetic reader (methyl-CpG-binding domain protein 2 v2 variant), essential for self-renewal and maintenance of these cells [111], suggesting an additional indirect role of diet on epigenetic information.
Histone acetylation mainly depends on the levels of acetyl-CoA, which is produced from glucose, acetate, ethanol, or FAO [52]. By neutralizing the positive histone charges, this epigenetic modification hampers DNA-histone association and induces chromatin opening and transcription. An increase in glycolysis-derived acetyl-CoA has been shown to be critical to maintaining the pluripotency of human and mouse SCs [115] and promoting their differentiation into regulatory T cells [116].
Histone acylation includes the addition of acyl groups derived from glycolysis (histone lactylation and succinylation), FA and protein metabolism (histone crotonylation), food additives (sodium benzoate-derived histone benzoylation), fasting-, exercise- and ketogenic diet-induced FA and ketogenic amino acid demolition (histone butyrylation and β-hydroxybutyrylation). Histone β-hydroxybutyrate (β-HB) marks active promoters of starvation-responsive genes [117] and is responsible for the protective effect on neurodegeneration, NSC maintenance [81], and small intestine crypt homeostasis [118], by inhibiting histone deacetylases (HDACs) and favoring histone and non-histone acetylation [81].
Histone homocysteinylation (Hcy), which may be driven by increased homocysteine levels in fetal brains, has been shown to modify gene expression and contribute to NTC defects [33]. In SCs, the availability of nutrients, including glucose, also regulates intracellular levels of UDP-GlcNAc, leading to mono-glycosyl-acylation (O-GlcNAcylation) of histones as well as of cellular proteins within the cytosol, mitochondria and nucleus, thus coupling metabolic signals to gene expression, self-renewal and differentiation [119].
Histone ADP-ribosylation requires the addition of mono- or poly-ADP-ribose (PARylation) residues derived from NAD+ to histones. Aging and DNA damage, as well as ROS and HFD, can induce poly(ADP-ribose) polymerase (PARP) enzymes and hence affect chromatin structure and gene expression [52].
Epigenetic modifications of DNA and histones may also take place non-enzymatically, through the addition of acetyl, methyl, and electrophilic groups. The methylglyoxal (MGO) group, derived from increased glycolysis, leads to the formation of advanced glycation end products (AGEs), which are associated with aging, diabetes, and cancer, and are responsible for modifications of nucleosomes, chromatin, and gene expression [120]. Sirtuins mediate the removal of MGO adducts and play an essential role in the maintenance of chromatin integrity. A similar effect can be mediated by ketogenic diet-induced acetoacetate, which inhibits the formation of AGEs by inhibiting MGO synthesis, thus affecting chromatin status and overall health [121].
The intracellular level of certain metabolites can also drive the elimination of epigenetic tags from histones and from DNA. Histone acetylation, for example, is removed by HDACs regulated by metabolites such as β-HB, derived from FAO or ketogenesis. In mouse ISCs, ketone bodies mediated by HDAC1 inhibition increase ISC self-renewal, differentiation, and regenerative capacity [122]. Activation of glycolytic metabolism leads to decreased NAD+ levels, thus reducing the NAD+-dependent histone deacetylase activity of SIRT1, leading to the consequent increase in H4K16 acetylation and satellite cell differentiation [39]. Reduced levels of acetyl-CoA due to HFD have also been associated with the reduction of white adipose tissue in mice [123]. Intracellular alpha-ketoglutarate, which can be derived from glucose and glutamine catabolism, contributes to the maintenance of the ESC pluripotency by promoting histone/DNA demethylation. Similarly, succinate induces differentiation by inhibiting histone demethylases [124]. In squamous cell carcinoma, EpdSCs were found dependent on extracellular serine to maintain elevated H3K27me3 levels, switching to de novo serine synthesis during serine starvation. This metabolic process has been shown to functionally activate α-ketoglutarate-dependent dioxygenases responsible for removing the repressive histone modification and activating cell differentiation and tumor growth inhibition [10].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23158108
References
- Kari Hemminki; Asta Försti; Meriem Khyatti; Wagida A. Anwar; Mohsen Mousavi; Cancer in immigrants as a pointer to the causes of cancer. European Journal of Public Health 2014, 24, 64-71, 10.1093/eurpub/cku102.
- Richard Doll; Richard Peto; The Causes of Cancer: Quantitative Estimates of Avoidable Risks of Cancer in the United States Today. JNCI: Journal of the National Cancer Institute 1981, 66, 1192-1308, 10.1093/jnci/66.6.1192.
- Paul Brennan; George Davey-Smith; Identifying Novel Causes of Cancers to Enhance Cancer Prevention: New Strategies Are Needed. JNCI Journal of the National Cancer Institute 2021, 114, 353-360, 10.1093/jnci/djab204.
- Shuji Ogino; Jonathan A. Nowak; Tsuyoshi Hamada; Danny A. Milner; Reiko Nishihara; Insights into Pathogenic Interactions Among Environment, Host, and Tumor at the Crossroads of Molecular Pathology and Epidemiology. Annual Review of Pathology: Mechanisms of Disease 2019, 14, 83-103, 10.1146/annurev-pathmechdis-012418-012818.
- Carla Almendáriz-Palacios; Darrell D. Mousseau; Christopher H. Eskiw; Zoe E. Gillespie; Still Living Better through Chemistry: An Update on Caloric Restriction and Caloric Restriction Mimetics as Tools to Promote Health and Lifespan. International Journal of Molecular Sciences 2020, 21, 9220, 10.3390/ijms21239220.
- Laura C. D. Pomatto-Watson; Monica Bodogai; Oye Bosompra; Jonathan Kato; Sarah Wong; Melissa Carpenter; Eleonora Duregon; Dolly Chowdhury; Priya Krishna; Sandy Ng; et al. Daily caloric restriction limits tumor growth more effectively than caloric cycling regardless of dietary composition. Nature Communications 2021, 12, 1-17, 10.1038/s41467-021-26431-4.
- E Morselli; M C Maiuri; M Markaki; E Megalou; A Pasparaki; K Palikaras; A Criollo; L Galluzzi; S A Malik; I Vitale; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death & Disease 2010, 1, e10-e10, 10.1038/cddis.2009.8.
- Valter D. Longo; Maira Di Tano; Mark P. Mattson; Novella Guidi; Intermittent and periodic fasting, longevity and disease. Nature Aging 2021, 1, 47-59, 10.1038/s43587-020-00013-3.
- Francesca Pistollato; Tamara Yuliett Forbes-Hernandez; Ruben Calderón Iglesias; Roberto Ruiz; Maria Elexpuru Zabaleta; Irma Dominguez; Danila Cianciosi; Josè L. Quiles; Francesca Giampieri; Maurizio Battino; et al. Effects of caloric restriction on immunosurveillance, microbiota and cancer cell phenotype: Possible implications for cancer treatment. Seminars in Cancer Biology 2020, 73, 45-57, 10.1016/j.semcancer.2020.11.017.
- Sanjeethan C. Baksh; Pavlina K. Todorova; Shiri Gur-Cohen; Brian Hurwitz; Yejing Ge; Jesse S. S. Novak; Matthew T. Tierney; June Dela Cruz-Racelis; Elaine Fuchs; Lydia W. S. Finley; et al. Extracellular serine controls epidermal stem cell fate and tumour initiation. Nature Cell Biology 2020, 22, 779-790, 10.1038/s41556-020-0525-9.
- Arieh Moussaieff; Matthieu Rouleau; Daniel Kitsberg; Merav Cohen; Gahl Levy; Dinorah Barasch; Alina Nemirovski; Shai Shen-Orr; Ilana Laevsky; Michal Amit; et al. Glycolysis-Mediated Changes in Acetyl-CoA and Histone Acetylation Control the Early Differentiation of Embryonic Stem Cells. Cell Metabolism 2015, 21, 392-402, 10.1016/j.cmet.2015.02.002.
- Massimiliano Cerletti; Young C. Jang; Lydia W.S. Finley; Marcia C. Haigis; Amy J. Wagers; Short-Term Calorie Restriction Enhances Skeletal Muscle Stem Cell Function. Cell Stem Cell 2012, 10, 515-519, 10.1016/j.stem.2012.04.002.
- Ömer H. Yilmaz; Pekka Katajisto; Dudley Lamming; Yetis Gültekin; Khristian E. Bauer-Rowe; Shomit Sengupta; Kivanc Birsoy; Abdulmetin Dursun; V. Onur Yilmaz; Martin Selig; et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 2012, 486, 490-495, 10.1038/nature11163.
- Victoria A. Rafalski; Elena Mancini; Anne Brunet; Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. Journal of Cell Science 2012, 125, 5597-5608, 10.1242/jcs.114827.
- Francis Blokzijl; Joep De Ligt; Myrthe Jager; Valentina Sasselli; Sophie Roerink; Nobuo Sasaki; Meritxell Huch; Sander Boymans; Ewart Kuijk; Pjotr Prins; et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538, 260-264, 10.1038/nature19768.
- Jesse S.S. Novak; Sanjeethan C. Baksh; Elaine Fuchs; Dietary interventions as regulators of stem cell behavior in homeostasis and disease. Genes & Development 2021, 35, 199-211, 10.1101/gad.346973.120.
- Rolf Bjerkvig; Berit B. Tysnes; Karen S. Aboody; Joseph Najbauer; A. J. A. Terzis; The origin of the cancer stem cell: current controversies and new insights. Nature Cancer 2005, 5, 899-904, 10.1038/nrc1740.
- Umesh Prasad Yadav; Tashvinder Singh; Pramit Kumar; Praveen Sharma; Harsimrat Kaur; Sadhana Sharma; Sandeep Singh; Santosh Kumar; Kapil Mehta; Metabolic Adaptations in Cancer Stem Cells. Frontiers in Oncology 2020, 10, 1010, 10.3389/fonc.2020.01010.
- Fidelia B. Alvina; Arvin M. Gouw; Anne Le; Cancer Stem Cell Metabolism. Adv. Exp. Med. Biol. 2021, 1311, 161-172, 10.1007/978-3-030-65768-0_12.
- Vivian Lu; Irena J. Roy; Michael A. Teitell; Nutrients in the fate of pluripotent stem cells. Cell Metabolism 2021, 33, 2108-2121, 10.1016/j.cmet.2021.09.013.
- Ilias Nikolits; Sabrina Nebel; Dominik Egger; Sebastian Kreß; Cornelia Kasper; Towards Physiologic Culture Approaches to Improve Standard Cultivation of Mesenchymal Stem Cells. Cells 2021, 10, 886, 10.3390/cells10040886.
- Manuel Doblado; Kelle H Moley; Glucose metabolism in pregnancy and embryogenesis. Current Opinion in Endocrinology, Diabetes & Obesity 2007, 14, 488-493, 10.1097/med.0b013e3282f1cb92.
- Chao Sun; Jin Shang; Yuan Yao; Xiaohong Yin; Minghan Liu; Huan Liu; Yue Zhou; O‐Glc NA cylation: a bridge between glucose and cell differentiation. Journal of Cellular and Molecular Medicine 2016, 20, 769-781, 10.1111/jcmm.12807.
- Xiaoyong Yang; Xiaoyong Yang Kevin Qian; Protein O-GlcNAcylation: emerging mechanisms and functions. Nature Reviews Molecular Cell Biology 2017, 18, 452-465, 10.1038/nrm.2017.22.
- Jilan Liu; Xianyun Qin; Dongfeng Pan; Bin Zhang; Feng Jin; Amino Acid-Mediated Metabolism: A New Power to Influence Properties of Stem Cells. Stem Cells International 2019, 2019, 1-9, 10.1155/2019/6919463.
- Lon J. Van Winkle; Rebecca Ryznar; One-Carbon Metabolism Regulates Embryonic Stem Cell Fate Through Epigenetic DNA and Histone Modifications: Implications for Transgenerational Metabolic Disorders in Adults. Frontiers in Cell and Developmental Biology 2019, 7, 300, 10.3389/fcell.2019.00300.
- Yuki Taya; Yasunori Ota; Adam C. Wilkinson; Ayano Kanazawa; Hiroshi Watarai; Masataka Kasai; Hiromitsu Nakauchi; Satoshi Yamazaki; Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science 2016, 354, 1152-1155, 10.1126/science.aag3145.
- Baubak Shamim; John A. Hawley; Donny M. Camera; Protein Availability and Satellite Cell Dynamics in Skeletal Muscle. Sports Medicine 2018, 48, 1329-1343, 10.1007/s40279-018-0883-7.
- Francesco Bifari; Sissi Dolci; Emanuela Bottani; AnnaChiara Pino; Marzia Di Chio; Stefania Zorzin; Maurizio Ragni; Raluca Georgiana Zamfir; Dario Brunetti; Donatella Bardelli; et al. Complete neural stem cell (NSC) neuronal differentiation requires a branched chain amino acids-induced persistent metabolic shift towards energy metabolism. Pharmacological Research 2020, 158, 104863, 10.1016/j.phrs.2020.104863.
- Brandon J. Gheller; Jamie E. Blum; Esther W. Lim; Michal K. Handzlik; Ern Hwei Hannah Fong; Anthony C. Ko; Shray Khanna; Molly E. Gheller; Erica L. Bender; Matthew S. Alexander; et al. Extracellular serine and glycine are required for mouse and human skeletal muscle stem and progenitor cell function. Molecular Metabolism 2020, 43, 101106, 10.1016/j.molmet.2020.101106.
- Michael S Kilberg; Naohiro Terada; Jixiu Shan; Influence of Amino Acid Metabolism on Embryonic Stem Cell Function and Differentiation.. Advances in Nutrition 2016, 7, 780S-9S, 10.3945/an.115.011031.
- Marie Clémot; Rafael Sênos Demarco; D. Leanne Jones; Lipid Mediated Regulation of Adult Stem Cell Behavior. Frontiers in Cell and Developmental Biology 2020, 8, 115, 10.3389/fcell.2020.00115.
- Qin Zhang; Baoling Bai; Xinyu Mei; Chunlei Wan; Haiyan Cao; Dan Li; Shan Wang; Min Zhang; Zhigang Wang; Jianxin Wu; et al. Elevated H3K79 homocysteinylation causes abnormal gene expression during neural development and subsequent neural tube defects. Nature Communications 2018, 9, 1-16, 10.1038/s41467-018-05451-7.
- Francesc R. Garcia-Gonzalo; Juan Carlos Izpisúa Belmonte; Albumin-Associated Lipids Regulate Human Embryonic Stem Cell Self-Renewal. PLOS ONE 2008, 3, e1384, 10.1371/journal.pone.0001384.
- Bo Wang; Xin Rong; Elisa N.D. Palladino; Jiafang Wang; Alan M. Fogelman; Martín G. Martín; Waddah A. Alrefai; David A. Ford; Peter Tontonoz; Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 2018, 22, 206-220.e4, 10.1016/j.stem.2017.12.017.
- Hui Zhang; Mehmet G. Badur; Ajit S. Divakaruni; Seth J. Parker; Christian Jäger; Karsten Hiller; Anne N. Murphy; Christian M. Metallo; Distinct Metabolic States Can Support Self-Renewal and Lipogenesis in Human Pluripotent Stem Cells under Different Culture Conditions. Cell Reports 2016, 16, 1536-1547, 10.1016/j.celrep.2016.06.102.
- Daniela Cornacchia; Chao Zhang; Bastian Zimmer; Sun Young Chung; Yujie Fan; Mohamed A. Soliman; Jason Tchieu; Stuart M. Chambers; Hardik Shah; Daniel Paull; et al. Lipid Deprivation Induces a Stable, Naive-to-Primed Intermediate State of Pluripotency in Human PSCs. Cell Stem Cell 2019, 25, 120-136.e10, 10.1016/j.stem.2019.05.001.
- Shohini Ghosh-Choudhary; Jie Liu; Toren Finkel; Metabolic Regulation of Cell Fate and Function. Trends in Cell Biology 2020, 30, 201-212, 10.1016/j.tcb.2019.12.005.
- James G. Ryall; Stefania Dell’Orso; Assia Derfoul; Aster Juan; Hossein Zare; Xuesong Feng; Daphney Clermont; Miroslav Koulnis; Gustavo Gutierrez-Cruz; Marcella Fulco; et al. The NAD+-Dependent SIRT1 Deacetylase Translates a Metabolic Switch into Regulatory Epigenetics in Skeletal Muscle Stem Cells. Cell Stem Cell 2015, 16, 171-183, 10.1016/j.stem.2014.12.004.
- Clifford Dl Folmes; Timothy J Nelson; Andre Terzic; Energy metabolism in nuclear reprogramming. Biomarkers in Medicine 2011, 5, 715-729, 10.2217/bmm.11.87.
- Athanasia Panopoulos; Oscar Yanes; Sergio Ruiz; Yasuyuki Kida; Dinh Diep; Ralf Tautenhahn; Aída Herrerías; Erika M Batchelder; Nongluk Plongthongkum; Margaret Lutz; et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Research 2011, 22, 168-177, 10.1038/cr.2011.177.
- S. Varum; O. Momčilović; C. Castro; A. Ben-Yehudah; J. Ramalho-Santos; C.S. Navara; Enhancement of human embryonic stem cell pluripotency through inhibition of the mitochondrial respiratory chain. Stem Cell Research 2009, 3, 142-156, 10.1016/j.scr.2009.07.002.
- Kenta Nishitani; Koji Hayakawa; Satoshi Tanaka; Extracellular glucose levels in cultures of undifferentiated mouse trophoblast stem cells affect gene expression during subsequent differentiation with replicable cell line-dependent variation. Journal of Reproduction and Development 2019, 65, 19-27, 10.1262/jrd.2018-083.
- Giuliano G. Stirparo; Agata Kurowski; Ayaka Yanagida; Lawrence E. Bates; Stanley E. Strawbridge; Siarhei Hladkou; Hannah T. Stuart; Thorsten E. Boroviak; Jose C. R. Silva; Jennifer Nichols; et al. OCT4 induces embryonic pluripotency via STAT3 signaling and metabolic mechanisms. Proceedings of the National Academy of Sciences 2021, 118, e2008890118, 10.1073/pnas.2008890118.
- Zhen Li; Jin Su; Mingming Sun; Jiaqi Song; Huanran Sun; Jun Fan; Guo Chen; Changliang Shan; Qi Qi; Shuai Zhang; et al. Octamer transcription factor-1 induces the Warburg effect via up-regulation of hexokinase 2 in non-small cell lung cancer. Molecular and Cellular Biochemistry 2021, 476, 3423-3431, 10.1007/s11010-021-04171-9.
- Marine Theret; Linda Gsaier; Bethany Schaffer; Gaetan Juban; Sabrina Ben Larbi; Michèle Weiss‐Gayet; Laurent Bultot; Caterina Collodet; Marc Foretz; Dominique Desplanches; et al. AMPK α1‐ LDH pathway regulates muscle stem cell self‐renewal by controlling metabolic homeostasis. The EMBO Journal 2017, 36, 1946-1962, 10.15252/embj.201695273.
- Qi Wang; Shudong Liu; Aihua Zhai; Bai Zhang; Guizhen Tian; AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation. Biological and Pharmaceutical Bulletin 2018, 41, 985-993, 10.1248/bpb.b17-00724.
- David B. Shackelford; Reuben J. Shaw; The LKB1–AMPK pathway: metabolism and growth control in tumour suppression. Nature Reviews Cancer 2009, 9, 563-575, 10.1038/nrc2676.
- Mohsen Sarikhani; Jessica C. Garbern; Sai Ma; Rebecca Sereda; Jeffrey Conde; Guido Krähenbühl; Gabriela O. Escalante; Aishah Ahmed; Jason D. Buenrostro; Richard T. Lee; et al. Sustained Activation of AMPK Enhances Differentiation of Human iPSC-Derived Cardiomyocytes via Sirtuin Activation. Stem Cell Reports 2020, 15, 498-514, 10.1016/j.stemcr.2020.06.012.
- Maria M. Mihaylova; Reuben J. Shaw; The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature Cell Biology 2011, 13, 1016-1023, 10.1038/ncb2329.
- Ki Wung Chung; Hae Young Chung; The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention. Nutrients 2019, 11, 2923, 10.3390/nu11122923.
- Ziwei Dai; Vijyendra Ramesh; Jason W. Locasale; The evolving metabolic landscape of chromatin biology and epigenetics. Nature Reviews Genetics 2020, 21, 737-753, 10.1038/s41576-020-0270-8.
- Masaki Igarashi; Leonard Guarente; mTORC1 and SIRT1 Cooperate to Foster Expansion of Gut Adult Stem Cells during Calorie Restriction. Cell 2016, 166, 436-450, 10.1016/j.cell.2016.05.044.
- David A. Guertin; David M. Sabatini; Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9-22, 10.1016/j.ccr.2007.05.008.
- Christopher L. Brooks; Wei Gu; How does SIRT1 affect metabolism, senescence and cancer?. Nature Cancer 2008, 9, 123-128, 10.1038/nrc2562.
- Yahyah Aman; Tomas Schmauck-Medina; Malene Hansen; Richard I. Morimoto; Anna Katharina Simon; Ivana Bjedov; Konstantinos Palikaras; Anne Simonsen; Terje Johansen; Nektarios Tavernarakis; et al. Autophagy in healthy aging and disease. Nature Aging 2021, 1, 634-650, 10.1038/s43587-021-00098-4.
- Laura García-Prat; Marta Martinez-Vicente; Eusebio Perdiguero; Laura Ortet; Javier Rodriguez-Ubreva; Elena Rebollo; Vanessa Ruiz-Bonilla; Susana Gutarra; Esteban Ballestar; Antonio L. Serrano; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37-42, 10.1038/nature16187.
- Theodore T. Ho; Matthew R. Warr; Emmalee R. Adelman; Olivia M. Lansinger; Johanna Flach; Evgenia V. Verovskaya; Maria E. Figueroa; Emmanuelle Passegué; Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205-210, 10.1038/nature21388.
- Natasha C. Chang; Autophagy and Stem Cells: Self-Eating for Self-Renewal. Frontiers in Cell and Developmental Biology 2020, 8, 138, 10.3389/fcell.2020.00138.
- Huize Pan; Ning Cai; Mo Li; Guang‐Hui Liu; Juan Carlos Izpisua Belmonte; Autophagic control of cell ‘stemness’. EMBO Molecular Medicine 2013, 5, 327-331, 10.1002/emmm.201201999.
- Nobuo N. Noda; Zheng Wang; Hong Zhang; Liquid–liquid phase separation in autophagy. Journal of Cell Biology 2020, 219, e202004062., 10.1083/jcb.202004062.
- Karyn E. King; Truc T. Losier; Ryan C. Russell; Regulation of Autophagy Enzymes by Nutrient Signaling. Trends in Biochemical Sciences 2021, 46, 687-700, 10.1016/j.tibs.2021.01.006.
- Owen A. Brady; Heba I. Diab; Rosa Puertollano; Rags to riches: Amino acid sensing by the Rag GTPases in health and disease. Small GTPases 2016, 7, 197-206, 10.1080/21541248.2016.1218990.
- In Hye Lee; Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Experimental & Molecular Medicine 2019, 51, 1-11, 10.1038/s12276-019-0302-7.
- Matthew R. Warr; Mikhail Binnewies; Johanna Flach; Damien Reynaud; Trit Garg; Ritu Malhotra; Jayanta Debnath; Emmanuelle Passegué; FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323-327, 10.1038/nature11895.
- Chong Chen; Yu Liu; Yang Liu; Pan Zheng; The axis of mTOR-mitochondria-ROS and stemness of the hematopoietic stem cells. Cell Cycle 2009, 8, 1158-1160, 10.4161/cc.8.8.8139.
- Francesca LiCausi; Nathaniel W. Hartman; Role of mTOR Complexes in Neurogenesis. International Journal of Molecular Sciences 2018, 19, 1544, 10.3390/ijms19051544.
- Yejing Ge; Jie Chen; Mammalian Target of Rapamycin (mTOR) Signaling Network in Skeletal Myogenesis. Journal of Biological Chemistry 2012, 287, 43928-43935, 10.1074/jbc.r112.406942.
- Xinxin Xiang; Jing Zhao; Geyang Xu; Yin Li; Weizhen Zhang; mTOR and the differentiation of mesenchymal stem cells. Acta Biochimica et Biophysica Sinica 2011, 43, 501-510, 10.1093/abbs/gmr041.
- Borzo Gharibi; Samira Farzadi; Mandeep Ghuman; Francis J. Hughes; Inhibition of Akt/mTOR Attenuates Age-Related Changes in Mesenchymal Stem Cells. Stem Cells 2014, 32, 2256-2266, 10.1002/stem.1709.
- Yao Liu; Xiaoxing Kou; Chider Chen; Wenjing Yu; Yingying Su; Yong Kim; Songtao Shi; Yi Liu; Chronic High Dose Alcohol Induces Osteopenia via Activation of mTOR Signaling in Bone Marrow Mesenchymal Stem Cells. Stem Cells 2016, 34, 2157-2168, 10.1002/stem.2392.
- Rogerio M. Castilho; Cristiane H. Squarize; Lewis A. Chodosh; Bart O. Williams; J. Silvio Gutkind; mTOR Mediates Wnt-Induced Epidermal Stem Cell Exhaustion and Aging. Cell Stem Cell 2009, 5, 279-289, 10.1016/j.stem.2009.06.017.
- Huiqiang Chen; Xianbao Liu; Han Chen; Jiang Cao; Ling Zhang; Xinyang Hu; Jiańan Wang; Role of SIRT1 and AMPK in mesenchymal stem cells differentiation. Ageing Research Reviews 2014, 13, 55-64, 10.1016/j.arr.2013.12.002.
- Katherine Blackmore; Weinan Zhou; Megan J Dailey; LKB1-AMPK modulates nutrient-induced changes in the mode of division of intestinal epithelial crypt cells in mice. Experimental Biology and Medicine 2017, 242, 1490-1498, 10.1177/1535370217724427.
- Yajing Liu; Junko Yamane; Akito Tanaka; Wataru Fujibuchi; Jun K. Yamashita; AMPK activation reverts mouse epiblast stem cells to naive state. iScience 2021, 24, 102783, 10.1016/j.isci.2021.102783.
- Miren Revuelta; Ander Matheu; Autophagy in stem cell aging. Aging Cell 2017, 16, 912-915, 10.1111/acel.12655.
- Kelsey L. Tinkum; Kristina M. Stemler; Lynn S. White; Andrew J. Loza; Sabrina Jeter-Jones; Basia M. Michalski; Catherine Kuzmicki; Robert Pless; Thaddeus S. Stappenbeck; David Piwnica-Worms; et al. Fasting protects mice from lethal DNA damage by promoting small intestinal epithelial stem cell survival. Proceedings of the National Academy of Sciences 2015, 112, 201509249-54, 10.1073/pnas.1509249112.
- Coralie Trentesaux; Marie Fraudeau; Caterina Luana Pitasi; Julie Lemarchand; Sébastien Jacques; Angéline Duche; Franck Letourneur; Emmanuelle Naser; Karine Bailly; Alain Schmitt; et al. Essential role for autophagy protein ATG7 in the maintenance of intestinal stem cell integrity. Proceedings of the National Academy of Sciences 2020, 117, 11136-11146, 10.1073/pnas.1917174117.
- Pu Xia; Xiao-Yan Xu; PI3K/Akt/mTOR signaling pathway in cancer stem cells: from basic research to clinical application.. American journal of cancer research 2015, 5, 1602-9, .
- Ataur Rahman; Subbroto Kumar Saha; Saidur Rahman; Jamal Uddin; Sahab Uddin; Myung-Geol Pang; Hyewhon Rhim; Ssang-Goo Cho; Molecular Insights Into Therapeutic Potential of Autophagy Modulation by Natural Products for Cancer Stem Cells. Frontiers in Cell and Developmental Biology 2020, 8, -, 10.3389/fcell.2020.00283.
- Zsolt Kovács; Brigitta Brunner; Csilla Ari; Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases. Nutrients 2021, 13, 2197, 10.3390/nu13072197.
- Daniela Liśkiewicz; Arkadiusz Liśkiewicz; Mateusz Grabowski; Marta Maria Nowacka-Chmielewska; Konstancja Jabłońska; Anna Wojakowska; Łukasz Marczak; Jarosław J. Barski; Andrzej Małecki; Upregulation of hepatic autophagy under nutritional ketosis. The Journal of Nutritional Biochemistry 2021, 93, 108620, 10.1016/j.jnutbio.2021.108620.
- Xing Fu; Meijun Zhu; Shuming Zhang; Marc Foretz; Benoit Viollet; Min Du; Obesity Impairs Skeletal Muscle Regeneration Through Inhibition of AMPK. Diabetes 2015, 65, 188-200, 10.2337/db15-0647.
- Mark F. McCarty; Nutraceutical and Dietary Strategies for Up-Regulating Macroautophagy. International Journal of Molecular Sciences 2022, 23, 2054, 10.3390/ijms23042054.
- Cord Naujokat; The “Big Five” Phytochemicals Targeting Cancer Stem Cells: Curcumin, EGCG, Sulforaphane, Resveratrol and Genistein. Current Medicinal Chemistry 2021, 28, 4321-4342, 10.2174/0929867327666200228110738.
- Mark F. McCarty; Lidianys Lewis Lujan; Simon Iloki Assanga; Targeting Sirt1, AMPK, Nrf2, CK2, and Soluble Guanylate Cyclase with Nutraceuticals: A Practical Strategy for Preserving Bone Mass. International Journal of Molecular Sciences 2022, 23, 4776, 10.3390/ijms23094776.
- Frank Madeo; Tobias Eisenberg; Federico Pietrocola; Guido Kroemer; Spermidine in health and disease. Science 2018, 359, eaan2788, 10.1126/science.aan2788.
- Na Li; Zhaoyu Du; Qiaoyan Shen; Qijing Lei; Ying Zhang; Mengfei Zhang; Jinlian Hua; Resveratrol Enhances Self-Renewal of Mouse Embryonic Stem Cells. Journal of Cellular Biochemistry 2017, 118, 1928-1935, 10.1002/jcb.25942.
- Anirban Roy Manju Ray; Anirban Roy; Kuladip Jana; Cancer Stem Cells, Wnt, Hedgehog and Notch Signaling, the Role of Dietary Phytochemicals: New Insights for Cancer Therapy. Translational Medicine 2013, 03, 1-5, 10.4172/2161-1025.1000e125.
- Yao Cheng; Jiachen Sun; Hui Zhao; Hongxing Guo; Jianying Li; Functional mechanism on stem cells by tea (Camellia sinensis) bioactive compounds. Food Science and Human Wellness 2022, 11, 579-586, 10.1016/j.fshw.2021.12.014.
- Larisa Ryskalin; Francesca Biagioni; Carla L. Busceti; Gloria Lazzeri; Alessandro Frati; Francesco Fornai; The Multi-Faceted Effect of Curcumin in Glioblastoma from Rescuing Cell Clearance to Autophagy-Independent Effects. Molecules 2020, 25, 4839, 10.3390/molecules25204839.
- Ji Wang; Chunying Wang; Gaofeng Bu; Curcumin inhibits the growth of liver cancer stem cells through the phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin signaling pathway. Experimental and Therapeutic Medicine 2018, 15, 3650-3658, 10.3892/etm.2018.5805.
- Nagarajan Maharajan; Karthikeyan Vijayakumar; Chul Ho Jang; Goang-Won Cho; Caloric restriction maintains stem cells through niche and regulates stem cell aging. Journal of Molecular Medicine 2019, 98, 25-37, 10.1007/s00109-019-01846-1.
- Sai Krupa Das; Priya Balasubramanian; Yasoma K. Weerasekara; Nutrition modulation of human aging: The calorie restriction paradigm. Molecular and Cellular Endocrinology 2017, 455, 148-157, 10.1016/j.mce.2017.04.011.
- Billel Benmimoun; Cédric Polesello; Lucas Waltzer; Marc Haenlin; Dual role for Insulin/TOR signaling in the control of hematopoietic progenitor maintenance in Drosophila. Development 2012, 139, 1713-1717, 10.1242/dev.080259.
- Mikhail V. Blagosklonny; Aging and Immortality: Quasi-Programmed Senescence and Its Pharmacologic Inhibition. Cell Cycle 2006, 5, 2087-2102, 10.4161/cc.5.18.3288.
- Maria M. Mihaylova; Chia-Wei Cheng; Amanda Q. Cao; Surya Tripathi; Miyeko D. Mana; Khristian E. Bauer-Rowe; Monther Abu-Remaileh; Laura Clavain; Aysegul Erdemir; Caroline A. Lewis; et al. Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell 2018, 22, 769-778.e4, 10.1016/j.stem.2018.04.001.
- Lisa Giovannelli; Beneficial effects of olive oil phenols on the aging process: Experimental evidence and possible mechanisms of action. Nutrition and Aging 2012, 1, 207-223, 10.3233/NUA-130016.
- Lucía Fernández Del Río; Elena Gutiérrez-Casado; Alfonso Varela-López; José M. Villalba; Olive Oil and the Hallmarks of Aging. Molecules 2016, 21, 163, 10.3390/molecules21020163.
- R. Santiago-Mora; A. Casado-Díaz; M. D. De Castro; J. M. Quesada-Gómez; Oleuropein enhances osteoblastogenesis and inhibits adipogenesis: the effect on differentiation in stem cells derived from bone marrow. Osteoporosis International 2010, 22, 675-684, 10.1007/s00198-010-1270-x.
- Shikhar Sharma; Theresa K. Kelly; Peter A. Jones; Epigenetics in cancer. Carcinogenesis 2009, 31, 27-36, 10.1093/carcin/bgp220.
- Oliver Hahn; Sebastian Grönke; Thomas Stubbs; Gabriella Ficz; Oliver Hendrich; Felix Krueger; Simon Andrews; QiFeng Zhang; Michael Wakelam; Andreas Beyer; et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biology 2017, 18, 1-18, 10.1186/s13059-017-1187-1.
- Mohsen Afarideh; Roman Thaler; Farzaneh Khani; Hui Tang; Kyra L. Jordan; Sabena M. Conley; Ishran M. Saadiq; Yasin Obeidat; Aditya S. Pawar; Alfonso Eirin; et al. Global epigenetic alterations of mesenchymal stem cells in obesity: the role of vitamin C reprogramming. Epigenetics 2020, 16, 705-717, 10.1080/15592294.2020.1819663.
- Sukanya Shyamasundar; Shweta P. Jadhav; Boon Huat Bay; Samuel Sam Wah Tay; S. Dinesh Kumar; Danny Rangasamy; S. Thameem Dheen; Analysis of Epigenetic Factors in Mouse Embryonic Neural Stem Cells Exposed to Hyperglycemia. PLOS ONE 2013, 8, e65945, 10.1371/journal.pone.0065945.
- Fei Chen; Linking metabolism to epigenetics in stem cells and cancer stem cells.. Seminars in Cancer Biology 2019, 57, iii–v., 10.1016/j.semcancer.2019.05.005.
- Diego Hernández-Saavedra; Rita S Strakovsky; Patricia Ostrosky-Wegman; Yuan-Xiang Pan; Epigenetic Regulation of Centromere Chromatin Stability by Dietary and Environmental Factors.. Advances in Nutrition 2017, 8, 889-904, 10.3945/an.117.016402.
- Michael Reid; Ziwei Dai; Jason W. Locasale; The impact of cellular metabolism on chromatin dynamics and epigenetics. Nature Cell Biology 2017, 19, 1298-1306, 10.1038/ncb3629.
- M. Perusina Lanfranca; J.K. Thompson; F. Bednar; C. Halbrook; Costas Lyssiotis; B. Levi; T.L. Frankel; Metabolism and epigenetics of pancreatic cancer stem cells. Seminars in Cancer Biology 2018, 57, 19-26, 10.1016/j.semcancer.2018.09.008.
- Aristeidis E. Boukouris; Sotirios D. Zervopoulos; Evangelos D. Michelakis; Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription. Trends in Biochemical Sciences 2016, 41, 712-730, 10.1016/j.tibs.2016.05.013.
- Emily A. Teslow; Cristina Mitrea; Bin Bao; Ramzi M. Mohammad; Lisa A. Polin; Greg Dyson; Kristen S. Purrington; Aliccia Bollig‐Fischer; Obesity‐induced MBD 2_v2 expression promotes tumor‐initiating triple‐negative breast cancer stem cells. Molecular Oncology 2019, 13, 894-908, 10.1002/1878-0261.12444.
- Özlem Altundag; Hande Canpinar; Betül Çelebi‐Saltik; Methionine affects the expression of pluripotency genes and protein levels associated with methionine metabolism in adult, fetal, and cancer stem cells. Journal of Cellular Biochemistry 2021, 123, 406-416, 10.1002/jcb.30180.
- Shuang Tang; Yi Fang; Gang Huang; Xiaojiang Xu; Elizabeth Padilla‐Banks; Wei Fan; Qing Xu; Sydney M Sanderson; Julie F Foley; Scotty Dowdy; et al. Methionine metabolism is essential for SIRT 1‐regulated mouse embryonic stem cell maintenance and embryonic development. The EMBO Journal 2017, 36, 3175-3193, 10.15252/embj.201796708.
- Nicholas D.E. Greene; Philip Stanier; Gudrun E. Moore; The emerging role of epigenetic mechanisms in the etiology of neural tube defects. Epigenetics 2011, 6, 875-883, 10.4161/epi.6.7.16400.
- Cara L. Green; Dudley W. Lamming; Luigi Fontana; Molecular mechanisms of dietary restriction promoting health and longevity. Nature Reviews Molecular Cell Biology 2021, 23, 56-73, 10.1038/s41580-021-00411-4.
- Arieh Moussaieff; Natalya M. Kogan; Daniel Aberdam; Concise Review: Energy Metabolites: Key Mediators of the Epigenetic State of Pluripotency. Stem Cells 2015, 33, 2374-2380, 10.1002/stem.2041.
- Min Peng; N. Yin; Sagar Chhangawala; Ke Xu; Christina S. Leslie; Ming O. Li; Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481-484, 10.1126/science.aaf6284.
- Zhigang Xie; Albert Jones; Jude Deeney; Seong Kwon Hur; Vytas A. Bankaitis; Inborn Errors of Long-Chain Fatty Acid β-Oxidation Link Neural Stem Cell Self-Renewal to Autism. Cell Reports 2016, 14, 991-999, 10.1016/j.celrep.2016.01.004.
- Christopher J. Terranova; Kristina M. Stemler; Praveen Barrodia; Sabrina L. Jeter-Jones; Zhongqi Ge; Marimar De La Cruz Bonilla; Ayush Raman; Chia-Wei Cheng; Kendra L. Allton; Emre Arslan; et al. Reprogramming of H3K9bhb at regulatory elements is a key feature of fasting in the small intestine. Cell Reports 2021, 37, 110044, 10.1016/j.celrep.2021.110044.
- Muhammad Abid Sheikh; Bright Starling Emerald; Suraiya Anjum Ansari; Stem cell fate determination through protein O-GlcNAcylation. Journal of Biological Chemistry 2021, 296, 100035, 10.1074/jbc.rev120.014915.
- Abdul Rouf Mir; Safia Habib; Moin Uddin; Recent advances in histone glycation: emerging role in diabetes and cancer. Glycobiology 2021, 31, 1072-1079, 10.1093/glycob/cwab011.
- Trine Salomón; Christian Sibbersen; Jakob Hansen; Dieter Britz; Mads Vandsted Svart; Thomas Schmidt Voss; Niels Møller; Niels Gregersen; Karl Anker Jørgensen; Johan Palmfeldt; et al. Ketone Body Acetoacetate Buffers Methylglyoxal via a Non-enzymatic Conversion during Diabetic and Dietary Ketosis. Cell Chemical Biology 2017, 24, 935-943.e7, 10.1016/j.chembiol.2017.07.012.
- Chia-Wei Cheng; Moshe Biton; Adam L. Haber; Nuray Gunduz; George Eng; Liam T. Gaynor; Surya Tripathi; Gizem Calibasi-Kocal; Steffen Rickelt; Vincent L. Butty; et al. Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell 2019, 178, 1115-1131.e15, 10.1016/j.cell.2019.07.048.
- Alessandro Carrer; Joshua L. D. Parris; Sophie Trefely; Ryan A. Henry; David C. Montgomery; Annmarie Torres; John M. Viola; Yin-Ming Kuo; Ian A. Blair; Jordan L. Meier; et al. Impact of a High-fat Diet on Tissue Acyl-CoA and Histone Acetylation Levels. Journal of Biological Chemistry 2017, 292, 3312-3322, 10.1074/jbc.m116.750620.
- Bryce W. Carey; Lydia W. S. Finley; Justin R. Cross; C. David Allis; Craig B. Thompson; Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2014, 518, 413-416, 10.1038/nature13981.
This entry is offline, you can click here to edit this entry!