1. Virulence Factors of S. aureus
The virulence factors of
S. aureus can be divided into the following categories: (1) secreted virulence factors, including toxins and superantigens, the main function of which is to disrupt host cell membranes and induce target cell lysis and inflammation [
26]; (2) extracellular enzymes, the main function of which is to break down host molecules for nutrition, promote bacterial survival and dissemination, etc. [
26]; (3) surface proteins of
S. aureus, whose main functions are adhesion, invasion, and immune escape [
27], and (4) pathogen-associated molecular patterns (PAMPs), which promote inflammatory responses [
28].
1.1. Secreted Virulence Factors
S. aureus can secrete a variety of enzymes and virulence factors that affect the immune system, leading to immune system dysregulation and the proliferation of auto-reactive T cells, as well as the development or progression of chronic autoimmune diseases. Virulence factors of
S. aureus include pore-forming toxins (PFTs) [
29], phenol-soluble modulins (PSMs) [
30], exfoliative toxins (ETs) [
31], and superantigens (SAgs) [
32] that activate different types of immune cells and cause several different inflammatory and infectious diseases.
1.1.1. PFTs
PFTs are important virulence factors secreted by bacteria that lead to cell lysis by forming pore structures in eukaryotic cell membranes. PFTs exert their toxic effects mainly by altering the permeability of cell membranes, leading to cell death [
29]. However, the disruption of cell permeability is often preceded by the release of cytokines and the activation of intracellular protein kinases. PFTs include α-hemolysin (Hla) [
33], β-hemolysin (Hlb), γ-hemolysin (Hlg) [
34,
35], α-toxin [
36], and Panton–Valentine leukocidin (PVL) [
37].
Haemolysin is a pore-forming toxin, also known as a membrane-disrupting toxin. Haemolysin is a substance that lyses red blood cells and releases hemoglobin, a sensitive, complementary fixed antibody that binds specifically to the antigen type of the red blood cell. This antibody can be produced by stimulation of the surface antigen and can cause red blood cells to lyse and release hemoglobin [
38]. Haemolysin includes Hla, Hlb, and Hlg.
Hla can induce the formation of small pores in the cell membrane, leading to the rapid release of K
+ ions as well as inducing the production of interleukin-1β (IL-1β), IL-6, and IL-8 [
33,
39,
40,
41]. Activated endothelial and epithelial cells induce the activation of caspase-1 and the production of NLRP3-inflammasome through the release of nitric oxide (NO), leading to extracellular Ca
2+ influx, the production of pro-inflammatory cytokines, and the pyroptosis of monocytes. Ca
2+ influx also activates caspase-12, activates caspase-3, and induces apoptosis. Hla also acts on Toll-like receptor (TLR) 3/4 and mediates necroptosis [
41,
42,
43,
44,
45,
46].
Hlb and Hlg can promote inflammasome formation containing procaspase-1, ASC, and NLRP3. Inflammasome activation promotes the release of IL-1β and IL-18. Activated caspase-1 can also cleave GSDMD to form the N-terminal cleavage product of GSDMD, and these together induce pyroptosis [
34,
35].
- 2.
-
α-Toxin
α-Toxin is a lecithin enzyme that breaks down lecithin, which is an important component of cell membranes. Therefore, α-toxin can damage the cell membrane of many cells, causing hemolysis, tissue necrosis, and vascular endothelial cell damage, increasing vascular permeability, and causing edema. α-Toxin activates macrophages and induces the activation of Th1 via CXCL-10. Th1 cells express interferon-γ (IFN-γ) in AD. α-Toxin induces early phagosomes (Rab5, Rab22b) to form autophagosomes (ILC3, Rab7), which induces cellular autophagy [
36]. α-Toxin also activates RIPK3, caspase-8, and caspase-12, mediating necroptosis and apoptosis. α-Toxin acts in AD, granulomatous polyangiitis (GPA), pneumonia, chronic sinusitis, sepsis, and other diseases [
47,
48,
49,
50,
51,
52,
53].
- 3.
-
PVL
PVL is a pore-forming toxin produced by
S. aureus that causes leukocyte destruction [
37]. PVL can induce the lysis of macrophages and neutrophils, leading to cell death [
54,
55]. PVL activates receptor-interacting serine threonine kinase 1 (RIPK1), RIPK3, and mixed-lineage kinase-like protein (MLKL) through tumor necrosis factor receptor 1 (TNFR1) and TLR3/4, forming a complex that leads to necroptosis. PVL can also lead to apoptosis. PVL also stimulates K
+ efflux, NLRP3 inflammasome production, and caspase-1 activation, leading to pyroptosis [
35,
56,
57,
58,
59,
60,
61]. PVL causes pneumonia through the above cell death mechanisms [
58,
62,
63,
64,
65,
66].
1.1.2. PSMs
PSMs are a family of amphipathic alpha-helical peptides found in
Staphylococci [
30]. PSMs contribute to biofilm structure and the propagation of biofilm-associated infections. PSMs include PSMα, PSMβ, and PSMγ.
PSMα can induce necroptosis in atopic dermatitis and pneumonia [
5,
67,
68,
69,
70]. PSMα induces the release of IL-1α and IL-36α from keratinocytes and induces the release of the pro-inflammatory cytokine IL-17, which mediates the skin inflammatory response to cause
S. aureus infection [
5]. PSMα induces the activation of keratinocytes and neutrophils, resulting in a series of pro-inflammatory responses (including cytokine production, leukocyte activation, and neutrophil chemotaxis) [
71,
72].
PSMα also activates and phosphorylates MLKL, as well as increases the expression and secretion of lactate dehydrogenase. Through MLKL and lactate dehydrogenase, PSMα can induce neutrophil necroptosis. The necroptosis of cells is the main pathological manifestation of
S. aureus pneumonia [
68].
- 2.
-
PSMβ
PSMβ has an important role in the formation of biofilms. PSMβ activates and induces neutrophil aggregation through formyl-peptide receptor 2 (FPR2), which induces an inflammatory response [
73]. In addition to the role of surfactant, PSMβ can also lyse erythrocytes and destroy them. However, in addition to the common properties of PSM, the unique role of PSMβ in
S. aureus itself has not yet been investigated [
30].
- 3.
-
PSMγ (δ Toxin)
The S. aureus δ toxin is a member of the PSM family. The δ toxin is cytolytic to neutrophils and erythrocytes.
The δ toxin is cytolytic to neutrophils and erythrocytes. The staphylococcal δ-toxin promotes allergic skin disease in mice by inducing mast cell degranulation [
74]. The δ toxin is an enterotoxin that is cytotoxic and increases cellular cAMP expression to inhibit water absorption in the ileum by altering the concentration of Na
+ and Cl
− in the mucosa, causing diarrhea [
75].
1.1.3. Proteases
ETs are extremely specific serine proteases secreted by
S. aureus. ETs can play a role in AD [
31]. ETs are the primary toxins that play a role in staphylococcal scalded skin syndrome (SSSS), an abscessing skin disease [
76].
ETs specifically recognize and hydrolyze the cell adhesion molecule desmoglein 1 (Dsg1), causing the dissociation of keratinocytes in human and animal skin and promoting skin infection by
S. aureus. Three distinct ET subtypes (ETA, ETB, and ETD) were identified in
S. aureus [
77].
- 2.
-
Serine Protease-Like Proteins (Spls)
Spls play a role in T cells and result in asthma [
78]. Spls recognize and hydrolyze desmoplastic proteins in the superficial skin layer, inducing skin peeling and blister formation [
79,
80,
81,
82,
83]. Spls trigger IgE antibody responses in most asthmatics. Peripheral blood T cells produce Th 2 cytokines after Spls stimulation. Therefore, Spls are considered to be triggering allergens in the allergic airway response to
S. aureus [
78]. T cells of CF patients produced more TH2 cytokines after stimulation by Spls [
84]. Increased IgE concentrations of Spls were detected in the serum of CF patients relative to healthy controls [
84].
- 3.
-
Staphopain B (SspB)
SspB is a human strain of
S. aureus that secretes papain-like proteases, and SspB has bacterial virulence. SspB can activate macrophages and lead to apoptosis [
85]. SspB may contribute to the recruitment of host cells, including immunomodulatory pDCs and/or macrophages, which contribute to the initiation and maintenance of a chronic inflammatory state by
S. aureus [
86].
1.1.4. SAgs
SAgs are a class of antigenic substances composed of bacterial exotoxins and retroviral proteins. They bind to most T cells and provide signals for T cell activation. SAgs are highly efficient T-cell mitogens and manipulate the host immune system. They can directly activate T lymphocytes, triggering the release of a large number of pro-inflammatory cytokines, such as IFN-γ, IL-2, and tumor necrosis factor (TNF) [
32,
87,
88]. SAgs include
Staphyloccucal enterotoxins (SEs) and toxic shock syndrome toxin 1 (TSST-1).
SEs include SEA, SEB, SEC, SEG, SHE, SEI, SEM, SEO, and SEQ (
Table 2). SEA and SEB can play a role in Th1, Th2, and Th22 cells and induce apoptosis. In nasal polyposis, SEB can induce IL-21 expression, and IL-21 also induces differentiation of Th17 [
89,
90]. SEA can lead to AD and asthma. SEB can cause AD, asthma, and chronic sinusitis. SEC can also lead to AD and cancer [
91,
92,
93]. SEA serves as a potent stimulant of PBMCs and induces the release of large amounts of cytokines and chemokines through the Src, ERK, and STAT pathways [
94]. SEB, SEG, SHE, SEI, SEM, SEO, and SEQ can also lead to food poisoning [
95,
96,
97,
98,
99,
100].
Table 2. The mechanisms and characteristics of different types of SEs.
- 2.
-
TSST-1
TSST-1 is a bacterial SAg produced and secreted by
S. aureus. TSST-1 can activate CD4
+ T cells to produce large amounts of cytokines and lead to a systemic toxic response. In AD, TSST-1 can lead to B lymphocytes and keratinocytes [
13,
50].
1.1.5. Secreted Enzymes (Exoenzymes) and Effectors
In addition to toxins,
S. aureus secretes many virulence factors with proenzymatic effects. These proenzymatic virulence factors can be broadly classified into two types: cofactors, which are used to activate host enzymes, and enzymes that lyse and destroy host cells and tissues. Secreted enzymes (exoenzymes) and cofactors act on different substances with different specific mechanisms of action, but their main function is to break down host cells and tissues to obtain nutrients for their growth, reproduction, and propagation [
26]. Secreted enzymes and effectors include EsxA, EsxB, coagulase (Coa), nuclease (Nuc), and adenosine synthase (AdsA), and staphopain (SspB).
EsxA and EsxB are small acidic proteins secreted by the early-secretion antigen-6 secretion system (ESS) as potential T-cell antigens of
S. aureus. ESS is the basic virulence factor of
S. aureus. EsxA and EsxB mediate the release of
S. aureus from host cells, which can cause apoptosis [
101,
102].
- 2.
-
Coa
Coa is an enzyme produced by
S. aureus. Coa has thrombospondin-like activity and coagulates plasma treated with citric or oxalic acid. Coa can induce coagulation with vascular hemophilia factor binding protein [
103]. Coa can lead to apoptosis [
104].
- 3.
-
Nuc and AdsA
Nuc can disrupt biofilms by breaking down extracellular DNA (eDNA) as well as mediating the escape of
S. aureus from the NET. The NET is an innate immune defense mechanism by means of which invading pathogens are removed [
105,
106]. Moreover, Nuc can degrade DNA in abscesses or NETs.
The degradation product, nucleotide monophosphate, can be a substrate for the synthesis of another adenylate, synthase A (AsdA). AdsA degrades DNA into deoxyadenosine, which induces the apoptosis of macrophages around NETs by caspase-3 activation. Thus, AsdA can promote the survival of
S. aureus [
107]. Nuc and AdsA can induce bacteraemia and nephrapostasis [
108].
- 4.
-
Extracellular Adhesion Protein (Eap)
Eap is the substance that mediates the adhesion of bacteria to host cells. In the early stages of infection,
S. aureus adheres and colonizes through the expression of Eap, secondary to infection. Eap can act on T cells and lung epithelial cells. It can also work in psoriasis and CF [
109].
1.2. Surface Proteins of S. aureus (Cell Wall-Anchored (CWA) Proteins)
Invasion of organs and tissues by S. aureus from the blood stream requires not only immune evasion but also adhesion. Biofilm formation is an important way for S. aureus to maintain infection. Adhesion, proliferation, and detachment are the main processes of biofilm formation.
Surface proteins of
S. aureus constitute a range of virulence factors, known as CWA proteins. CWA proteins play a key role in the adhesion phase. The sorting signal of CWA proteins is responsible for covalently coupling proteins to peptidoglycan (PGN) [
110]. There are 24 different CWA proteins on the surface of
S. aureus, including microbial surface components recognizing adhesive matrix molecules (MSCRAMMs), the near iron transporter (NEAT) motif protein family, three-helical bundle motif protein A, G5-E repeat family, legume-lectin, and cadherin-like domain protein [
111]. CWA proteins play an important role in extracellular matrix (ECM) adhesion, host cell invasion, and immune response evasion. Therefore, targeting CWA proteins with vaccines can counteract
S. aureus infections [
112].
1.2.1. MSCRAMM
MSCRAMMs are a series of proteins with similar amino acid sequences and very similar structures and functions. MSCRAMMs consist of two folded subdomains similar to IgG, and the two folded subdomains are adjacent to each other. MSCRAMM plays an important role in tissue invasion during
S. aureus infection, including enabling
S. aureus to adhere to and invade host cells and tissues, evade host immune attack, and induce biofilm formation [
110]. Therefore, targeting the MSCRAMM protein is an alternative immunotherapeutic direction for the therapy of
S. aureus infections [
113].
1.2.2. Staphylococcal Protein A (SpA)
SPA is a protein that forms the cell wall of
S. aureus. It was found that SpA can form a complex with human immunoglobulin, and the complex formed by the two can induce the necrosis of various immune cells in the body [
114]. SpA can induce apoptosis and cause osteomyelitis [
115,
116,
117,
118].
The second immunoglobulin-binding protein (Sbi) belongs to SpA and consists of four triple-helix bundles arranged in tandem. Two of the triple-helix bundles bind similarly to those of SpA and IgG. Sbi non-covalently binds lipoteichoic acid (LTA), which then binds to the cell envelope and helps S. aureus to evade attack by the host immune system.
S. aureus Sbi can induce IL-33 production from keratinocytes in AD [
119]. IL-33 can induce itch. IL-33 is a type 2 cytokine and a major regulator of chronic itch [
120]. Sbi-inducible expression of IL-33 causes pruritus in AD [
121]. It is therefore a virulence factor that promotes type 2 immune response.
1.3. PAMPs
PAMPs are bacterial-specific structures that typically activate TLRs, which act as neutrophil activators [
28]. PAMPs of
S. aureus include triacyl lipopetides, diacyl lipoproteins, and LTA.
1.3.1. Triacyl Lipopetides and Diacyl Lipoproteins
Triacyl lipopetides and diacyl lipoproteins are components of the
Staphylococcal cell wall that provide structural integrity and protection against virulence organisms from the host immune system. Triacyl lipopetides and diacyl lipoproteins can be distinguished by subtle differences in TLR 1 and TLR 6 interactions with TLR2 [
122]. Triacyl lipopetides and diacyl lipoproteins can induce activation of the TLR 2 and NLRP3 inflammasome. NLRP3 activation induces pyroptosis. Lipopetides and diacyl lipoproteins can induce the activation of TLR 2 and the NLRP3 inflammasome [
35,
69,
123]. Triacyl lipopetides and diacyl lipoproteins can make use of sepsis [
69,
124].
1.3.2. LTA
LTA is an adhesion affinity agent associated with surface adhesion and a modulator of the bacteria’s own cell wall lysis enzymes (Muramidases). LTA is released from the bacteria after rupture and death. LTA can induce the phenomenon of passive immune killing. LTA promotes macrophage activation and the expression of IL-1β and IL-18 [
125]. LTA activates neutrophils and macrophages and leads to pyroptosis. LTA can also work in food poisoning [
23,
125,
126,
127].
1.3.3. PGN
PGN is the main structural component of the cell wall [
128], a glycopolymer that maintains the shape of the bacteria. PGN is also a key factor in bacterial recognition by the host immune system [
129].
PGN induces the activation of keratinocytes, Langerhans cells (LCs), mast cells, macrophages, CD4
+T cells, Th1 cells, and microglia, and induces the release of several cytokines, including TNF-α, IL-1, and IL-4 [
17,
130,
131,
132,
133,
134,
135,
136,
137,
138,
139]. PGN causes autophagy, which drives AD, sepsis, pneumonia, and psoriasis [
17,
130,
131,
132,
133,
140,
141]
S. aureus contains large amounts of SpA, which evades phagocytosis and uptake by phagocytes by binding to immunoglobulin (Ig) molecules and then covering the bacterial surface. Peptidoglycan can promote the B-cell superantigen activity of SpA [
142].
The cells involved in S. aureus infection and inflammation include keratinocytes, T cells (helper T cells, ILCs), macrophages, DCs, mast cells, neutrophils, eosinophils, and basophils.
2.1. Keratinocytes/Epithelial Cells
Keratinocytes are the main cellular components that make up the epidermis. S. aureus expresses PSMα, which is a group of secreted virulence peptides.
In AD,
S. aureus accumulates in the lysosomes of keratinocytes and induces IL-1α secretion via TLR9 [
152].
S. aureus expresses PSMα acting on keratinocytes, induces IL-1α and IL-36α release from keratinocytes via myd88 signaling, induces γδt cell and ILC3 production of IL-17, and promotes neutrophil infiltration [
5]. In addition,
S. aureus promotes IL-36α secretion from keratinocytes and promotes IgE production and allergic inflammatory response [
70].
S. aureus promotes IL-1β expression by keratinocytes and induces skin regeneration through keratinocyte-dependent IL-1R-MyD88 signaling [
153].
S. aureus Sbi promotes IL33 secretion by keratinocytes, and IL-33 promotes type 2 immune responses [
119,
154]. IL-33 also has an important role in pruritus [
155].
S. aureus diacylated lipoprotein is a TLR2 and TLR6 ligand, which activates keratinocytes via TLR2-TLR6 heterodimers to produce TSLP, which promotes T(H)2-type inflammation and thus AD [
156]. The keratinocyte-expressed TSLP acts directly on TRPA1-positive sensory neurons, triggering intense pruritus-induced scratching [
157].
In nasal polyp tissue,
S. aureus can directly induce the release of the epithelial cell-derived cytokines TSLP and IL-33 by binding to TLR 2, thereby potentially propagating the expression of type 2 cytokines IL-5 and IL-13 in nasal polyp tissue [
158,
159,
160,
161].
2.2. Helper T Cells (Th Cells)
2.2.1. T Helper 1 (Th1) Cells
Th1 cells are CD4
+ cells that mainly secrete IL-2, IFN-γ, and TNF-β (tumor necrosis factor β), which are involved in regulating cellular immunity, assisting in the differentiation of cytotoxic T cells, mediating cellular immune responses, and participating in delayed hypersensitivity reactions. IFN-γ, IL-2, and TNF-β can drive inflammatory responses [
162].
S. aureus infection can induce Th1-type inflammatory responses in different diseases. Enterotoxin B (SEB) induces Th1 activation in chronic sinusitis; α-toxin promotes Th1 activation via CXCL10 in AD [
48,
49,
93,
163]. Activated Th1 cells release the cytokine IFN-γ. IFN-γ is a driver of chronic inflammation in the chronic phase of AD, and the overexpression of IFN-γ can lead to recurrent inflammation and pruritus, causing lichenoid degeneration of the skin [
13,
164]. In contrast, psoriasis is also a chronic disease mediated by Th1 cytokines, and IL-12 mediates the differentiation of Th1 cells [
165].
2.2.2. Th2 Cells
Th2 cells (which are CD4+ T cells) are important cells in the type 2 inflammatory pathway. th2 secretes IL-4, IL-5, and IL-13 and stimulates type 2 immunity, as evidenced by the production of high levels of IgE and eosinophils. Type 2 immunity is a specific immune response that includes both innate and adaptive immunity and contributes to the formation of an immune barrier on the mucosal surface to a clear response to pathogens [
166]. Th2 cells are formed by the induced differentiation of Th0 cells by IL-4 produced by basophils, eosinophils, mast cells, natural killer cells (NK), or already differentiated Th2 cells.
In AD infection with
S. aureus, keratinocytes can express TSLP, IL33 [
119,
154], and IL-19 [
167], which can induce the Th2 expression of IL-4, IL-10, IL-13, and IL-31, causing an inflammatory response. IL-4 and IL-31 play important roles in itchiness [
158,
159,
167].
S. aureus was found to induce the expression of IL-8, IL-19, and IL-22, which induced increased expression of Th2 cytokines [
131,
167,
168].
S. aureus PGN action on LCs induces Th2 cells in the skin via CCL17, and it induces IL-18 stimulation of CD4
+ T cells to produce IL-4 to induce Th2 cytokine expression [
130,
131].
Staphylococcal α-toxin mediates the expression of Th2 cytokines IL-4 and IL-13 through STAT6, leading to increased keratin-forming cell death [
169].
It was found that antigen-specific regulatory T (Treg) cells can also induce Th2 cell proliferation [
170].
S. aureus PGN action on LCs induces Th2 cells in the skin via CCL17, and it induces the IL-18 stimulation of CD4
+ T cells to produce IL-4, which induces Th2 cytokine expression [
130,
131]. In addition,
staphylococcal α-toxin mediates the expression of Th2 cytokines IL-4 and IL-13 through STAT6, leading to increased keratin-forming cell death [
169]. SEB can also act on eosinophils, monocytes, and Treg cells to induce Th2 cytokine expression and induce the Th2 proliferation and secretion of IL-10 to aggravate skin inflammation [
170,
171]. In asthma, eosinophils induce the activation of Th2 and express IL-5 in conjunction with basophils [
172]. In chronic sinusitis, Th2 cytokines can activate B cells, release IgE, and indirectly mediate eosinophil inflammation [
7]. Th2 cytokines can reduce filamentous protein expression in keratinocytes, further exacerbating the disruption of skin barrier function [
173]. Th2 expresses IL-4, IL-10, IL-13, and IL-31, inducing epidermal thickening, sensitization, inflammation, and pruritus [
174,
175]. Th2 also reduces the expression of AMP, HBD-2, HBD-3, filamentous polymerase, and epidermal proteins, further promoting the proliferation of
S. aureus and exacerbating flora imbalance. The activation of keratinocytes increased the expression of endogenous serine proteases, induced inflammatory responses and tissue damage, and released various cytokines. Serine protease V8 and the serine protease strip toxin cleave corneal adhesion proteins, including DSG-1, leading to increased desquamation [
158,
159].
2.2.3. Th17 Cells
Th17 is a newly discovered subpopulation of T cells that secrete IL-17, which is important in autoimmune diseases and the body’s defense response. Transforming growth factor β (TGF-β), IL-6, IL-23, and IL-21 play active roles in the differentiation and formation of Th17 cells, while IFN-γ, IL-4, cytokine signaling (IFN-γ), IL-4, suppressor of cytokine signaling 3 (Socs3), and IL-2 inhibit its differentiation [
176].
Th17 cells have a crucial role in host defense. Dysregulated Th17 responses mediate various autoimmune and inflammatory diseases. IL-6, TGF-β, and IL-23, secreted by macrophages and DCs, induce the activation and differentiation of Th17. Th17 cells are recruited into the skin and interact with keratinocytes and fibroblasts to promote epidermal tissue repair [
164]. Th17 cells also express IL-17, IL-22, and IL-26. IL-17 can coordinate local tissue by upregulating pro-inflammatory cytokines and chemokines (including IL-1β, IL-6, TNF-α, granulocyte-macrophage colony-stimulating factor (GM-CSF), KC/CXCL1, MCP-1/CCL2, MIP-2/CXCL2, MCP-3/CCL7, and MIP-3α/CCL20, and matrix metal proteinases (MMPs)), enabling the migration of activated T cells through the ECM [
177,
178,
179].
S. aureus triggers a strong Th17-type response in skin effector T cells, inducing Th17 to produce IL-17A, IL-17F, and IL-22 [
180].
S. aureus promotes keratinocyte expression of IL1β, promotes differentiation of Th17, and promotes skin inflammation [
181]. SEB induces IL-21 secretion by Follicular helper T (Tfh), induces differentiation of Th17, and acts in nasal polyp disease [
89,
90].
In septic arthritis caused by
S. aureus infection,
S. aureus activates immune cell-specific macrophages and DCs to release pro-inflammatory mediators, such as TNF-α, IL-1β, IL-6, and IL-21, which induce RORγt, RORγt, and CD4 T cells to differentiate into Th17 cells [
182,
183,
184]. Th17 cells produce the pro-inflammatory cytokine IL-17, which directly stimulates arthritic inflammation by binding to receptors on immune cells, stimulating the production of more pro-inflammatory cytokines, chemokines, and other inflammatory mediators, including NO and MMPs, while TNF-α, IL-1β, and IL-6 upregulate MMPs, which can promote cartilage degradation and enhance joint destruction [
185,
186].
IL-17 activates macrophages to express TNF-α and IL-1β as well as fibroblasts to produce IL-6, IL-8, and MMPs; it also stimulates blood endothelial cells to produce chelators and the p38 MAPK-dependent expression of VCAM-1 and ICAM-1, which contribute to immune cell evasion from blood to tissue [
180]. In mouse studies, ICAM-1 was upregulated on endothelial cells in the diseased skin of CD18 (β2-integrin) (CD18hypo) mice, whereas in vivo
S. aureus extracellular adhesion protein prevented T cell vascular migration to the inflamed skin of CD18 (β2-integrin) (CD18hypo) mice but did not inhibit their proliferation and activation [
109]. IL-17 also promoted epithelial cell stimulation by IL-8 and granulocyte colony-stimulating factor (G-CSF), induced neutrophil migration and activation, and synergistically enhanced the induction of TNF-α. IL-22 not only triggers a pro-inflammatory response, but also inhibits terminal differentiation of keratinocytes and induces Th2-type inflammation [
168].
In psoriasis, IL-17 can activate macrophages to express TNF-α and IL-1β, thereby inducing fibroblast activation [
180]. In inflammatory areas of the skin, Th17 can increase the inflammatory response of the skin, and IL-17A can strongly induce IL-19 expression in keratinocytes. IL-19 also induces the expression of Th2 cytokines [
167,
187].
2.2.4. Tregs
Tregs, characterized by an expression of the forkhead transcription factor FOXP3 and IL-2R α chain CD25, play a central role in maintaining tolerance to self and preventing an overexcited inflammatory response to infection [
188].
S. aureus induces the proliferation of effector memory T (Tem) cells and the activation of Treg cells and Th17 responses through cutaneous LCs [
189,
190]. Th17 and Treg cells antagonize each other, and Th17/Treg cell imbalance can trigger or exacerbate the disease process in AD [
191]. After superantigenic stimulation of SEs, Treg cells lose their immunosuppressive activity, and a Treg induces Th2 proliferation and the secretion of IL-10 to aggravate the skin inflammatory response [
170].
2.2.5. Tfh Cells
Tfh cells are a specialized subset of CD4
+ T cells located in B cell follicles that induce B and T cell interactions and release cytokines to promote germinal center (GC) formation, promote the differentiation of GC B cells into memory B cells or plasma cells, and drive the maturation of high-affinity antibodies [
192]. The Tfh cells express CXC chemokine receptor 5 (CXCR5) [
193], ICOS [
194], programmed death-1 (PD-1) [
195], cytotoxic T lymphocyte antigen 4 (CTLA-4) [
196], B-cell lymphoma-6 (BCL-6) [
197], and IL-21 [
90].
In
S. aureus-infected chronic rhinosinusitis with nasal polyposis (CRSwNP), Tfh can migrate into lymphoid aggregates (lymphoid aggregates resemble germinal centers) and interact with B cells to induce the proliferation and differentiation of naive B cells to plasma cells [
89]. Tfh is also involved in the B cell-mediated immune response induced by the
S. aureus vaccine [
198]. In nasal polyposis, IL-21 expression was increased after SEB stimulation. IL-21 can also induce the differentiation of Th17 [
89,
90].
However, there has been minimal research on this aspect of the mechanism of Tfh in S. aureus infection, and future research in this direction may allow the mechanism of S. aureus infection to be further deciphered and allow for more comprehensive counseling for clinical treatment.
2.2.6. Th9 Cells
Recently, researchers have identified another novel and unique population of immunomodulatory cells. Th9 cells were initially thought to be a subpopulation of Th2 cells that could produce IL-9 [
199]. Th9 cells have been shown to act on many cell types associated with asthma, including T cells, B cells, mast cells, eosinophils, neutrophils, and epithelial cells, and therefore may be important in the pathophysiology of allergic asthma [
200]. Moreover, Th9 has been found to be a major factor in regulating immunity to autoimmune diseases [
201] and tumor immunity [
202].
There are few studies on this aspect of the mechanism of S. aureus infection promoting Th9-type inflammatory response, which may be a direction for further research.
2.3. ILCs
ILCs are lymphocytes that do not express the diverse antigen receptor types expressed on T and B cells [
203]. ILCs comprise a heterogeneous population of immune cells that maintain barrier function and can initiate a protective or pathological immune response in response to infection [
204].
ILCs can be divided into ILC1, ILC2, and ILC3 types. In cell-mediated effector immunity, ILC1, ILC2, and ILC3 are involved in type 1, type 2, and type 17 immune responses, respectively [
205].
S. aureus and
S. aureus muramyl dipeptide (MDP) can activate ILC2 and promote type 2 immune response [
161]. In AD, PSMα induces the production of ILC3, which is involved in the mediation of skin inflammation [
5]. In patients with AD infection with
S. aureus, the expansion of ILC3 is involved in mediating skin inflammation [
13].
However, little is known about the relationship between S. aureus infection and ILCs. This could be another direction in the study of the mechanism of S. aureus infection in the future.
2.4. Macrophages
Macrophages are cellular components of the innate immune system and are present in almost all tissues, contributing to immunity, repair, and homeostasis [
206]. When
S. aureus infects humans, epithelial cells recognize the invading
S. aureus through pathogen recognition receptors (PRRs), which induce the production of pro-inflammatory cytokines and chemokines, leading to the recruitment and activation of phagocytes, including GM-CSF, G-CSF, IL-1β, IL-6, and IL-8 [
8].
Phagocytosis of
S. aureus triggers the TLR2-dependent signaling and activation of the NLRP3 inflammasome, leading to the recruitment of ASC and the activation of cystathione-1, causing cells to release cytokines IL-1β and IL-18 and inducing apoptosis [
207,
208].
S. aureus was found to survive and replicate in macrophages, which deliver nutrients to lysosomal-engulfing
S. aureus to promote bacterial growth, and promote bacterial persistence during infection by limiting reactive oxygen species (ROS) and RNS production by macrophages through lipoic acid synthesis [
209,
210].
2.5. DCs
DCs are the most powerful specialized antigen-presenting cells in the body and are highly efficient in the uptake, processing, and presentation of antigens. Immature DCs have a strong migratory capacity and mature DCs can effectively activate initial T cells and are central to the initiation, regulation, and maintenance of the immune response. They are usually found in small numbers in contact with external skin and their immature forms can be found in the blood. When activated, they move to the lymphoid tissue to interact with T and B cells to stimulate and control the appropriate immune response.
S. aureus activates macrophages and DCs to release pro-inflammatory mediators, such as TNF-α, IL-1β, IL-6, and IL-21, which induce RORγt to produce IL-17A, IL-17F, and IL-21 and induce CD4 T cells to differentiate into Th17 cells [
182,
183,
184].
TSLP released by keratinocytes in AD is a potent activator of DCs, triggering the production of Th2-attracting chemokines, such as CCL17/TARC and CCL22/MDC, and inducing Th2 differentiation through upregulation of these cells by OX40L, which activates Th2 cells [
9]. In experiments with psoriatic mice,
S. aureus Eap was found to disrupt cell–cell contacts between T cells and DCs in vitro, blocking T cell extravasation into inflamed skin to inhibit psoriasis [
109].
LCs are the major DCs in the normal epidermis, and DCs are the major antigen-presenting cells [
211]. Epidermal exposure to
S. aureus induces the proliferation of effector memory T (Tem) cells and limited Treg cell activation and Th17 responses, and LCs directly interact with
S. aureus via the pattern recognition receptor langerin (CD207), which interacts directly with
S. aureus [
189,
190]. LCs express TLRs that recognize bacterial and viral products, and the TLR2-mediated transduction of
S. aureus-derived signals is severely impaired in LCs with AD skin [
212]. The DC-mediated blockade of human T cell activation and proliferation, PVL, targets DCs to blunt CD4
+ T lymphocyte activation and kills DCs, leading to impaired T cell responses and increased infection [
213].
2.6. Mast Cells
Mast cells are granulocytes that disintegrate to release granules and the substances in the granules. Such granules in the blood contain heparin, histamine, and 5-hydroxytryptamine, which can drive tachyphylactic allergic reactions (inflammation) in tissues, especially in asthma. Mast cells are the main effector cells of inflammation, and mast cells act as antigen-presenting cells and induce Th1 and Th2 cell development.
In patients with AD,
S. aureus infection leads to an increase in the number of mast cells, which proliferate within mast cells and mediate Th1 cell development as well as the development of chronic inflammation, leading to the release of Th1 cytokines and the upregulation of IFN, manifested as edema within the lamina propria [
214,
215]. In mast cells, PGN from
S. aureus stimulates mast cells in a TLR2-dependent manner, producing TNF-α, IL-4, IL-5, IL-6, and IL-13, and
S. aureus also triggers TNFα and IL-8 release by binding to CD48 [
132,
133].
The δ toxin induces mast cell degranulation, which is dependent on intracellular PI3K activation and free calcium ion influx. δ toxin-induced mast cell degranulation differs from conventional IgE cross-linking in that this action does not require the presence of an antigen. IgE enhances δ toxin-induced mast cell degranulation, promotes IgE and IL-4 production, and leads to skin inflammation. However, AD is a chronic inflammatory skin disease caused by mast cells, leading to immunoglobulin-E (lgE)-mediated hypersensitivity [
74,
147].
S. aureus-expressed SAgs can also trigger mast cell degranulation via IgE and FcεR, and SEs may also trigger direct histamine release from mast cell degranulation via unknown receptors [
10]. In asthma, the airways are hyperreactive, and mast cells release histamine and various cytokines that attract the accumulation of eosinophils, causing epithelial cell damage and respiratory distress [
216,
217].
Although mast cells may help clear the infection, S. aureus may use mast cells to evade detection and immune clearance.
2.7. Neutrophils
Neutrophils are a type of myeloid leukocyte and some of the major responders in acute inflammation. Activated neutrophils mediate inflammation by synthesizing and secreting cytokines, chemokines, leukotrienes, and prostaglandins. Neutrophils synthesize and secrete the chemokine CXCL8 to recruit more neutrophils and express IL-1, IL-6, IL-12, TGF-β, and TNF-α, reactivating neutrophils and other cells of the immune system [
218].
S. aureus can recruit and activate neutrophils at the site of infection [
219]. In AD and pneumonia, PSMα can induce the expression of IL-1α and IL-36 and induce neutrophil death, leading to disease exacerbation [
67,
68]. γδ T cells mediate IL-17 responses and induce neutrophil recruitment, pro-inflammatory cytokines IL-1α, IL-1β, and TNF, and host defense peptides, while rapid neutrophil recruitment enhances
S. aureus colonization in the skin [
11]. IL-17 also promotes IL-8 and G-CSF stimulation of epithelial cells, induces neutrophil migration and activation, synergistically enhances TNF-α induction, and increases antimicrobial peptide (AMP) production [
180].
Neutrophils induce IL-20 expression, which contributes to psoriasis, wound healing, and anti-inflammatory effects [
165].
S. aureus LTA promotes the expression of neutrophil factors TNF-α and IL-8. IL-8 also attracts neutrophils to accumulate in the intestine, leading to the activation and attraction of polymorphonuclear leukocytes (PMN), causing an inflammatory response in the intestine during food poisoning [
23]. The α toxin produces IL-1β via TLR2, NOD2, FPR1, and ASC/NLRP3 inflammasome induced by neutrophils, and IL-1β can induce thymic stromal lymphopoietin and contribute to abscess formation [
178,
220].
S. aureus α toxin produces IL-1β via TLR2, NOD2, FPR1, and ASC/NLRP3 inflammasomes induced by neutrophils expressing IL-1β [
220]. In GPA, the activation of neutrophils, by expressing neutrophil extracellular trap products (NET-derived products), plays a role in the disease [
221].
2.8. Eosinophils (Eos)
Eosinophils are considered to be the effector cells associated with infection and the cause of tissue damage. Eosinophils can express a range of ligand receptors that play a role in cell growth, adhesion, chemotaxis, degranulation, and intercellular interactions. Eosinophils synthesize, store, and secrete cytokines, chemokines, and growth factors. Eosinophils can function as antigen-presenting cells and can regulate the immune system [
222].
In AD,
S. aureus produces exotoxins that can amplify the allergic response by directly activating other immune cells, such as eosinophils. Eosinophil inflammation can be induced by SEB. Eosinophils lead to epidermal damage, tissue swelling, and inflammatory cell recruitment through the release of toxic mediators, including eosinophil cationic protein (ECP), major basic proteins, eosinophil-derived neurotoxins, and eosinophil peroxidase (EPO) [
223,
224]. In addition, activated eosinophils can promote antimicrobial defense by releasing mitochondrial DNA associated with granule proteins [
12].
In asthma, eosinophils are not only involved in the release of granulins, lipid mediators, ROS, cytokines, and growth factors that trigger the Th2 response [
172]. The IL-5 cytokines of Th2, GM-CSF, and IL-3 induce the eosinophil response and eosinophil maturation. IL-3 and GM-CSF also induce eosinophil recruitment [
225,
226].
In CRSwNP,
S. aureus can also cause eosinophil inflammation by inducing IgE [
7].
2.9. Basophils
Basophils are a type of leukocyte that originate from bone marrow hematopoietic pluripotent stem cells that differentiate and mature in the bone marrow and enter the bloodstream. Basophils are important cells for
S. aureus respecting the virulence of AD and asthma [
3,
216].
S. aureus SAgs, including SEA, SEB, SEC, and TSST-1, also directly activate B lymphocytes and induce specific IgE-dependent mast cells and basophil degranulation to release histamine, further exacerbating AD and promoting adaptive cellular and humoral type 2 immunity [
13].
NOD2 and TLR2 ligands trigger basophil activation by interacting with dermal fibroblasts in AD-like skin inflammation [
227].
S. aureus was found to induce skin basophil aggregation and increase IL-4 expression. Basophil-derived IL-4 inhibited skin IL-17A production by TCRγδ+ cells and promoted
S. aureus infection of the skin. Basophils secrete IL-6 to promote Th17 responses and inhibit the IL-17A production of IL-4 via STAT6 inhibition of the IL-17A promoter to promote
S. aureus infection [
228], while Toll-like expressing receptor-expressing epidermal keratinocytes recognize invasion and respond to LTA by inducing the expression of cytokines such as TSLP, which leads to basophil recruitment and IL-4 production [
126].
2.10. B Cells
In inflammatory and neoplastic diseases, B cells play a regulatory role by secreting regulatory cytokines, such as IL-10, or by relying on the secretion of antibodies. B cells can act in concert with other immunomodulatory cells, such as Treg cells. TSST-1 can induce B cell apoptosis [
229]. LTA inhibits LPS-induced B cell proliferation by reducing ERK phosphorylation through the TLR2 signaling pathway [
230]. The
S. aureus-induced activation of Th2 triggers B cells to produce IgE in response to allergens and autoantigens, indirectly mediating eosinophil inflammation [
7]. Tfh and ILCs can also induce B cell differentiation [
89,
216].
In the disease response to S. aureus infection, there are many other cells involved besides those mentioned above. As a major human pathogen, S. aureus induces cell activation in various cell types, releases various cytokines, and causes apoptosis, which is important in S. aureus infections. However, the cellular mechanisms are not yet clear, including the role of Th9, Tfh, and ILCs in the pathogenesis of S. aureus. This may be another direction for studies on S. aureus infection in the future.
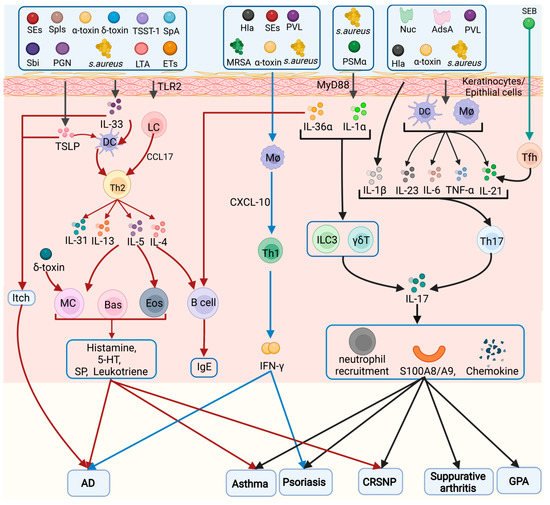
A summary of the inflammatory cells activated by S. aureus in several diseases is shown in Figure 1.
Figure 1. Inflammatory cell types in the pathogenesis of Staphylococcus aureus. Different virulence factors of S. aureus can induce activation of Tfh, Th1, Th2, Th9, and Th17 cells, which play a role in chronic sinusitis, AD, asthma, itch, psoriasis, septic arthritis, and CGD. Eos: eosinophils, Bas: basophils, MC: mast cells, Mø: macrophage.
This entry is adapted from the peer-reviewed paper 10.3390/toxins14070464