Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related death worldwide. This high mortality rate is due to the disease’s lack of symptoms, resulting in a late diagnosis. Biomarkers and treatment options for pancreatic cancer are also limited. In order to overcome this, new research models and novel approaches to discovering PDAC biomarkers are required. In this review, we outline the hereditary and somatic causes of PDAC and provide an overview of the recent genome wide association studies (GWAS) and pathway analysis studies. We also provide a summary of some of the systems used to study PDAC, including established and primary cell lines, patient-derived xenografts (PDX), and newer models such as organoids and organ-on-chip. These ex vitro laboratory systems allow for critical research into the development and progression of PDAC.
- pancreatic cancer
- GWAS
- genomics
- organoids
- cancer models
- Introduction
With a five-year survival rate of 9%, pancreatic cancer has one of the worst outcomes of all cancers. Due to its rapid progression and fatal outcome, long-term survivors are limited to those with resected early-stage tumours [1,2]. As 80% of pancreatic cancer patients are diagnosed after the disease has metastasized, most people diagnosed are ineligible for resection, the only curative treatment. It is the fourth leading cause of cancer-related death in the Western world, and by 2030 it is estimated that pancreatic cancer will surpass breast and colorectal cancer to become the second most fatal cancer in the United States [3]. The most common form of pancreatic cancer is pancreatic ductal adenocarcinoma (PDAC), which occurs in the exocrine pancreas, with the remaining 5% of cases in the endocrine pancreas [4]. Epidemiological factors, including smoking, obesity, type II diabetes mellitus and acute pancreatitis account for approximately 25% of cases of PDAC [5–8].
The mean survival time of patients who receive the surgery and adjuvant treatment is 11 to 23 months. Of the patients who are operated on, 60% relapse within 12 months; this is most likely due to micro-metastases which were not detected during the diagnostic computed tomography (CT) scan [9]. Approximately 25–30% of patients treated with chemotherapeutic drugs respond, however most eventually become resistant. Resistance mechanisms include deficiencies in drug uptake, alteration of drug targets, activation of DNA repair pathways, and resistance to apoptosis [10]. Gemcitabine is the mainstay of modern chemotherapy for pancreatic cancer [11]. FDA approval has also been granted for gemcitabine in combination with erlotinib and paclitaxel in 2005 and 2013 respectively [12]. The drug combination FOLFIRINOX (irinotecan, oxaliplatin, 5-fluorouracil, and leucovorin) was approved by the FDA in 2011.
- Genomic Variants of PDAC
In addition to epidemiological factors which account for 25% of cases, research into the genetic landscape of the disease, including familial cancer syndromes, inherited predisposition loci and somatic mutations is vital to identifying those at risk of developing the disease.
2.1. Familial Cancer Syndromes
Familial cancer syndromes including Peutz-Jegher Syndrome (PJS), pancreatitis, familial atypical multiple mole and melanoma syndrome (FAMMM), Lynch syndrome, Hereditary Breast and Ovarian Cancer (HBOC) syndrome and Familial adenomatous polyposis (FAP), account for approximately 5–10% of pancreatic cancers. Table 1 contains an outline of the diseases and syndromes associated with an increased risk of developing PDAC.
Table 1. Familial Cancer Syndromes associated with an increased risk of developing pancreatic ductal adenocarcinoma (PDAC). The table includes increased risk, genes associated with syndrome/disease, pathways associated with syndrome/disease and pathway function.
|
PJS1 |
Pancreatitis |
FAMMM2 |
Lynch Syndrome |
HBOC3 |
FAP4 |
|
|
Increased Risk |
132-fold |
69-fold |
13–22-fold |
8.6-fold |
3.5–10-fold |
4.5–6-fold |
|
Genes |
STK11/LKB11 |
PRSS1 SPINK1 CFTR |
CDNK2A |
MLH1 MSH2 MSH6 PMS2 |
BRCA1 BRCA2 PALB2 |
APC |
|
Pathways |
AMPK/mTOR |
Trypsin |
Retinoblastoma |
Mismatch repair |
Homologous recombination repair |
Wnt signalling |
|
Pathway Function |
Cell growth Polarity Metabolism |
Auto-activation of trypsin |
G1 to S-phase checkpoint |
Maintenance of genomic stability |
Repair of double-strand breaks in DNA |
Regulation of gene transcription |
1 Peutz-Jegher Syndrome; 2Familial atypical multiple mole and melanoma syndrome; 3Hereditary Breast and Ovarian Cancer syndrome; 4Familial adenomatous polyposis.
Peutz-Jegher Syndrome (PJS) is a rare autosomal dominant disease, characterised by gastrointestinal polyposis, mucocutaneous pigmentation, and cancer predisposition [13]. PJS increases the risk of several malignancies, including breast, pancreatic and gynaecological cancers [14]. Individuals with PJS have a 132-fold increased risk of developing PDAC [15]. It is caused by a mutation in STK11, also known as liver kinase B1 (LKB1). STK11/LKB1 is a serine/threonine protein kinase which drives many cell functions, including cell growth, regulation of metabolism and cell polarity, mainly through AMP-activated protein kinase/ mammalian target of rapamycin (AMPK/mTOR) signalling [16]. The most common STK11/LKB1 mutations are deletions or inactivating mutations. In a genetically engineered mouse model (GEMM) study by Helez et al.[17] STK11/LKB1 deletion resulted in defective acinar cell polarity, with abnormal cytoskeleton, loss of tight junctions, and progressive acinar degeneration. Deletion of STK11/LKB1 in the pancreas also resulted in the development of serous cystadenomas. Morton et al. [18] showed that the STK11/LKB1 deletion resulted in accelerated KRASG12D tumorigenesis, through decreased TP53 and p21 dependent growth arrest. These studies, along with others provide strong evidence for a tumour suppressor function for this gene [19].
Pancreatitis is the second most common hereditary cause of PDAC. Pancreatitis is an inflammatory disorder of the pancreas, caused by the premature activation or lack of inhibition of digestive enzymes. There are several forms of hereditary pancreatitis, including a gain of function mutation in serine-1 protease gene (PRSS1), which makes trypsinogen [20]. This gain of function mutation results in increased trypsinogen auto-activation, which triggers pancreatic self-digestion. Other genes associated with hereditary pancreatitis include SPINK1, a pancreatic secretory trypsin inhibitor and CFTR (cystic fibrosis transmembrane regulator) [21]. The chronic inflammation of the pancreas which characterises pancreatitis result in the presence of reactive oxygen species (ROS) in the pancreas [22]. These ROS, including nitric oxide and free radicals inhibit apoptosis, and can result in direct DNA damage, resulting in oncogenic mutations in genes such as KRAS, CDKN2A and TP53 [23,24]. Cytokines which are released in response to pancreatitis activate pancreatic stellate cell and result in the development of fibrosis, facilitating the development of PDAC [25–27]. People with chronic or hereditary pancreatitis have a 69-fold increased risk of pancreatic cancer [28].
An autosomal dominant disorder, familial atypical multiple mole and melanoma syndrome (FAMMM) is characterised by melanoma in more than one first- or second-degree relative, high total body mole count (often >50), and moles with certain histopathological features. The melanomas can arise from the atypical moles or de novo, superficially spreading melanoma and/or nodular melanoma [29]. Three original descriptions in different kindreds implicated germline mutations or microdeletions in cyclin-dependent kinase inhibitor 2A (CDNK2A), in particular the p16INK4a isoform, as causative for FAMMM [30]. FAMMM results in a 13 to 22-fold increased risk of PDAC. CDKN2A is also mutated in 90–95% of sporadic PDACs [31,32]. It inhibits cyclin dependent kinases 4/6 (CDK4/CDK6) and thereby activates the retinoblastoma (RB) family of proteins, which blocks the transition from G1 to S-phase [33]. It is mainly associated with the autosomal dominant familial melanoma, but patients also have an increased risk of PDAC [34]. By identifying individuals with an increased risk of developing PDAC from a family history, or families with a gene defect which results in PDAC, Vasen et al. [35] detected PDAC in 7.3% of the CDKN2A mutation carriers, by providing an annual Magnetic Resonance Imaging (MRI) scan, resulting in a resection rate 75% and an overall 5-year survival rate of 24%.
Lynch syndrome is also associated with an increased risk of colorectal cancer and PDAC [36]. It is caused by mutations in the mismatch repair genes (MMR), mainly MutL homolog 1 (MLH1), MutS homolog 2/6 (MSH2/MSH6) and PMS1 Homolog 2 (PMS2). The MMR maintains the integrity of the genome by repairing DNA replication errors [37]. Bi-allelic loss of MMR genes results in genomic instability, and an increase of unrepaired replication errors, particularly affecting repeats, such as microsatellites, termed microsatellite instability-high (MSI-H). MSI-high results in genome hypermutability, with a 100- to 1000- fold increase in mutations [38,39]. Individuals with Lynch Syndrome have 8.6-fold increased risk of PDAC [40].
Hereditary Breast and Ovarian Cancer syndrome is caused by mutations in the tumour suppressor genes BRCA1 and BRCA2. People with this syndrome have a 3.5–10-fold increased risk of developing PDAC [41]. Mutated variants of the Partner and Localiser of BRCA2 (PALB2) gene are also associated with a familial risk of PDAC [42]. PALB2 has a critical role in homologous recombination repair (HRR) and recruits BRCA2 and RAD51 to DNA breaks. Jones et al. [43] found that in 96 patients with PDAC, three had truncating mutations in the PALB2 gene, producing a stop codon, which was not present in 1084 healthy controls. Slater et al. [44] observed a similar prevalence of PALB2 mutations (3.7%) in a panel of 81 European patients with familial pancreatic cancer. A study in a 61-year-old patient with advanced localised PDAC, with a bi-allelic inactivation of PALB2, found treatment with Mitomycin C resulted in disease regression, and at the 3-year follow up, the patient remained asymptomatic. Studies also showed that patients with wild type PALB2 are Mitomycin C resistant [45].
Familial adenomatous polyposis (FAP), is a familial cancer syndrome which results in an increased risk of colorectal cancer [46,47]. It is characterised by colorectal polyps, due to a mutation in the adenomatous polyposis coli (APC) gene. APC acts to negatively regulate the Wnt signalling pathway [48]. The Wnt proteins stabilise cytosolic β-catenin, which associates with the transcriptional regulators T cell factor/lymphoid enhancer factor-1 family (TCF), thereby allowing the expression of Wnt-regulated genes [49]. Murine studies of colorectal cancer have found that mutations in the APC gene result in hyperproliferation of cells [50]. Individuals with FAP have 4.5 to 6-fold increased risk of PDAC [47].
2.2. Inherited Predisposition Loci
Recently, the landscape of pancreatic cancer has been redefined through gene expression and genetic diversity signatures identified using next generation sequencing (NGS) and genome wide association studies (GWAS). GWAS examines hundreds of thousands of variants, in thousands of individuals, to identify genotype-phenotype associations, and helps to identify risk factors for multifactorial diseases [51]. Through this, GWAS can enable the identification of people at risk of developing a disease, and also can be used for the examination of the biological underpinnings of a disease. GWAS enables the use of potential preventative measures for those who are identified as at risk, and also for the development of treatments for the disease. GWAS use single nucleotide polymorphisms (SNPs) which are single base pair changes in the genome. SNPs can occur in the gene, in both introns and exons which result in amino acid changes, different mRNA splicing and reduce the mRNA transcript stability [52]. SNPs can also occur in the transcriptional regulatory elements such as transcription factor binding sites, enhancers and promoters, resulting in altered mRNA expression [53].
Several PDAC GWAS studies have been performed over the past decade [54–59] and have identified common variants associated with risk of pancreatic cancer in European populations (Figure 1). Obazee et al. [60] used the PANDoRA dataset to validate a truncating BRCA2k336X (rs11571833) and pathogenic CHEK2I157T (rs17879961) variants. Both genes are critical in DNA repair and the maintenance of genomic stability. While the results of the GWAS have informed the genetic component of predisposition loci, it does not give a clear indication of the cause of PDAC. The use of complementary GWAS pathway analysis—a method of analysing this genomic data through sets defined by functional pathways—offers the potential of greater power for discovery and natural connections to biological mechanisms. Pathway analysis allows for the identification of causative SNPs whose individual effects may not be significant enough to be detected in GWAS [61]. Pathway analysis of PDAC GWAS SNP data has been previously performed [62,63]. Walsh et al.[63] performed a pathway-analysis based on meta-analysis of PDAC GWAS. Pathways associated with the development of the pancreas, including pancreas development and the regulation of beta cell development were among the pathways identified in these studies (Table 2).
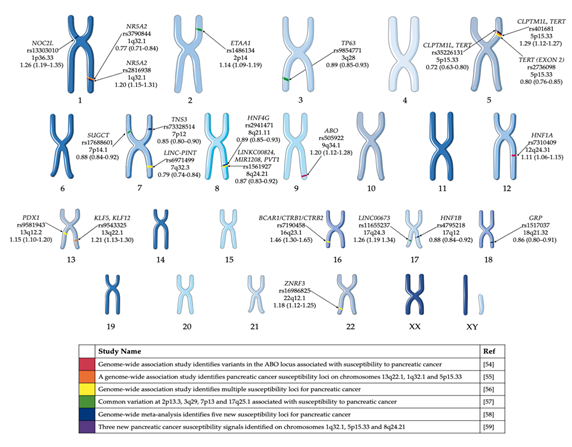
Figure 1. GWAS significant single nucleotide polymorphisms (SNPs) identified in pancreatic cancer cases of European ancestry. Highlighted GWAS SNP, closest gene, chromosome and odds ratio (95% confidence interval) [54–59]. This figure was created using Servier Medical Art templates, which have been modified. These images are licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com.
Table 2. Gene sets/pathways identified for risk of developing PDAC from pathway analysis studies [62,63].
|
Pathway/Gene Set |
Pathway Reference |
Study |
Pathway p-Value |
|
Maturity onset diabetes of the young |
KEGG |
[63] |
5.10 × 10−7 |
|
Regulation of Beta cell development |
REACTOME |
[63] |
1.92 × 10−6 |
|
Breast Cancer 17Q11 Q21 amplicon 1. |
NIKOLSKY |
[63] |
2.00 × 10−6 |
|
Role of EGF Receptor Transactivation by GPCRs in Cardiac Hypertrophy |
BIOCARTA |
[63] |
3.79 × 10−6 |
|
ATM Pearson Correlation Coefficient (PCC) Network 2. |
PUJANA |
[63] |
1.25 × 10−5 |
|
Pancreas development |
|
[62] |
2.0 × 10−6 |
|
Heliobacter pylori lacto/neolacto |
|
[62] |
1.6 × 10−5 |
|
Hedgehog |
|
[62] |
0.0019 |
|
Th1/Th2 immune response |
|
[62] |
0.019 |
|
Apoptosis |
|
[62] |
0.023 |
- Genes within amplicon 17q11-q21 identified in a copy number alterations study of 191 breast tumour samples. 2. Gene network transcripts whose expression positively correlated with ATM gene in normal tissues.
A recent study by Campa et al. [64] looked at the genetics of early onset pancreatic cancer (EOPC)—disease which occurs in those sixty-years or younger, and represents 20% of cases of PDAC. Four SNPs (rs7155613, rs2328991, rs4891017 and rs12610094) were found to be associated with EOPC (p < 1 × 10−4). Of the SNPs identified, rs2328991 at 13q22.3 was found to be significant in the replication dataset. The SNP is 57 kb from the 3′ UTR of the potassium channel tetramerization containing 12 gene (KCTD12) which has previously been implicated in gastrointestinal stromal tumours.
2.3. Somatic Mutations
Studies into the PDAC genome have shown that there are approximately 60 alterations per tumour; most of which are point mutations [65]. Activating mutations of KRAS are nearly universal, and inactivation of TP53, SMAD4 and CDKN2A occur at rates of >50% (Figure 2) [66].
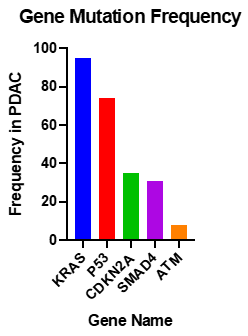
Figure 2. Most common somatic common gene mutations in PDAC [67,68].
KRAS is a molecular switch, when bound to GTP regulates cell proliferation, differentiation, apoptosis and cell signalling. The KRAS mutation is near universal in PDAC, with 94% of tumours possessing the mutation [68]. Activating point mutations in codon 12, 13, or 16, (most commonly G12D), result in reduced GTP hydrolysis. Cases with KRAS mutations at codon 61 give a favourable prognosis, as there is less ERK activation [69]. KRAS mutations in pancreatic cancer are believed to be the early events in neoplastic transformation [70]. Oncogenic KRAS is not sufficient to initiate the carcinogenesis process, this relies on the downstream activation of Raf-1, Rac, Rho or PI3K [71]. RAS proteins are modified by farnesyl transferase, an enzyme which adds 15-carbon farnesyl lipid to the carboxyl-terminal cysteine of RAS. This modification is shown to be essential for RAS membrane association and transformation [72]. A Phase III clinical trial of R115777, a selective inhibitor of farnesyl transferase combined with gemcitabine failed to show increased life expectancy in comparison to gemcitabine plus placebo [73]. Patients with KRAS mutations were associated with a median survival time of 17 months compared to 30 months for those without mutations [70]. Approximately 3% of PDAC cases are due to microsatellite instability or altered chromosome ploidy [74]. This is usually due to mutations in the MMR genes MSH2 and MSH6. Typically, KRAS is wild-type in cancers with these mutations.
CDKN2A, previously discussed as the causative gene of FAMMM, is inactivated in 95% of PDAC cases, by homozygous deletion, mutation of alleles or promoter hypermethylation resulting in gene silencing [75].
TP53 is the most frequently mutated gene in cancer [76]. It acts as a tumour suppressor, and has roles in apoptosis, genomic stability, inhibition of angiogenesis and arrest of cell growth [77]. It is mutated in 75% of PDAC tumours, mainly by point mutations [78]. TP53 controls cell cycle at the G1/S interface and plays a vital role in inducing programmed death in response to DNA damage [1]. Weissmueller et al. [79] found mutant TP53 induced platelet-derived growth factor receptor b (PDGFRb). Knockdown of PDGFRb in PDAC cell lines resulted in reduced invasion of the cells. Mutant TP53 inhibits p73, which represses PDGFRb. The study found that increased expression of PDGFRb in PDAC patient samples correlates with a worse outcome for patients.
SMAD4 is a transcription factor in TGFβ signalling pathway and is inactivated in 50% of advanced pancreatic cancers. It acts with TGFβ1 as a tumour suppressor to regulate pancreatic cell cycle arrest, and apoptosis, mediated by targets such as p21, which causes G1 cell cycle arrest [80]. Patients with biallelic deletion of SMAD4 more frequently had metastasis than those with wild type SMAD4 [81].
Whole genome sequencing of PDAC identified other genes which are frequently mutated in PDAC, such as ataxia telangiectasia mutated (ATM), a serine/threonine kinase with a role in DNA double strand break repair; and pathways including the TGFβ, the β-catenin and Notch pathways [58]. Recent data based on large-scale sequencing studies reported up to 18% of ATM mutations in PDAC cohorts [65,67,68,82,83].
- Models of PDAC Research
3.1. Established PDAC Cell Line Cultures
Two-dimensional, cell-based assays are an important tool, and have been the mainstay of cancer research for over 50 years. The first cell line (HeLa) was developed in 1950 from cervical carcinoma [84]. Cell lines are able to grow indefinitely, making them an easy to use, low cost, repeatable model, and thus important for both drug discovery, and proof-of-concept studies. While the usefulness of cell lines in cancer research is certain, their use as a clinical model is debatable [85]. Cell lines often undergo genetic modifications, including copy number variation and point mutations during passaging [86]. Cell lines tend to be homogeneous which does not represent the heterogeneous nature of PDAC tumours. Cell lines are often developed from late stage, aggressive tumours, so they cannot be used to model tumour progression [87].
PDAC cell lines recapitulate the genomic changes which lead to the development of the disease. The four most common mutations (KRAS, TP53, CDKN2A SMAD4) occurring in PDAC tumours are found in cell lines at similar percentages and PDAC cell lines also demonstrate the different phenotypes and genotypes which are found in PDAC subclasses. A commonly used PDAC cell line is BxPC3, developed from pancreatic adenocarcinoma of a 61-year old female. BxPC3 has TP53 mutations, a homozygous deletion in SMAD4, but is CDKN2A wild type and is the only KRAS wild type PDAC cell line. Other common PDAC cell lines include, PANC-1, developed from PDAC of a 56-year old male and MIAPaCa-2, developed from a PDAC of a 65-year old male, harbour mutations in KRAS and TP53, with homozygous deletion in CDKN2A and wild type SMAD4. Capan-1 was developed from a liver metastasis of a 40-year-old male with PDAC, and harbours mutations in KRAS, TP53, CDKN2A and SMAD4, and is the only PDAC cell line with a BRCA2 mutation. A detailed review from Deer et al. [88] provides an overview of the available information on the most commonly used PDAC cell lines.
Due to genomic drift, differences in cell culture procedures and media, in different labs may result in genotypic and phenotypic differences in the same cell line. Recently, Ben-David et al. [89] performed a full genomic characterisation of 27 different strains of the ER-negative breast cancer cell line MCF7. Changes were observed including differential activation of gene expression programs, morphology and proliferation. Drug sensitivity was shown to vary in the cell lines, with 75% of the drugs that were tested which strongly inhibited some of the MCF7 cell lines, were completely inactive in others.
Another issue with the use of established cell lines include cross contamination. Boonsta et al. [90] have identified two oesophageal adenocarcinoma cell lines which were contaminated, and have been used in 11 patents, and more than 100 published studies, leading to clinical trials. Horbach and Halffman [91] identified 32,755 articles reporting on research with misidentified cells, which in turn have been cited by over half a million papers. To overcome these issues, a number of journals require cell lines to verify before publishing a research paper. The method used to validate the cell lines are short tandem repeat (STR) profiling. These techniques were initially developed for forensic applications [92]. STR profiling compares microsatellite (2 to 7 base pairs) repeats at specific loci which are unique to each individual [93]. It is carried out by using commercially available PCR primers which are compared to size markers, allowing for a comparison of the lengths of the PCR products at each locus to the STR profile made from the original donor material [94].
Two-dimensional (2D) established cell line models have been standard method for cancer drug testing for many years, however, of late, the limitations of using established cell lines in 2D are being increasingly recognised. In actuality, 2D cell culture platforms often fail to recapitulate the physiology of tumours in vivo due to different cellular architecture, adherence structures and biochemical gradients.
3.2. Primary Cell Lines
Primary cell lines are an emerging tool for cancer research. These cell lines are derived from a patient tumour or biopsy, dissociated, and grown in vitro [95]. Primary cell lines are heterogeneous, and are at an early passage number, so are more representative of the original tumour [96]. Primary cell lines may allow for the development of personalised cancer therapy through the development of primary cell lines from patient tumour, and the function testing of chemotherapeutic drugs on the living cancer cells [97]. While primary cultures are more representative of the original patient tumour, there are several issues with PDAC primary cell lines—they are often difficult to establish, only grow for a limited number of passages, and often tumour cells are overgrown by stromal cells such as fibroblasts [96,97].
3.3. Organ-on-Chip
“Organ-on-chip” is a microfluidic chip containing multiple cell types which are joined by microchannels and can simulate the activities of entire organ-systems. These cancer models can be used to represent the tumour microenvironment, and can show cancer initiation and progression, observations of the interactions and signalling pathways of different cell types [98]. This model can be used to identify potential metastasis sites of the tumour, observed the impact of immune cells in cancer, and to determine the effects of cancer treatment on other organs [99,100].
Beer et al. [101] used HepaChip organ specific 3D cell culture chambers to culture PDAC cell lines PANC-1, BcPC3 and MIAPaCa-2 with the extracellular matrix protein collagen. The cells maintained the viability, morphological appearance, and growth characteristics of 3D spheroids when grown on a chip.
Using an organ-on-chip model, Nguyen et al. [102] studied tumour-endothelium in PDAC, which is a poorly vascularised cancer. PDAC 3D organotypic models were placed in a chamber next to endothelialised, perfused lumen. The study found that through the TGF-β receptor signalling pathway, activin-ALK7 allowed for endothelial ablation, where the PDAC cells invaded and removed the vascular endothelium, leaving tumour filled structures. These results were then validated in vivo.
3.4. Patient Derived Xenografts (PDX)
Patient derived xenografts (PDX) are another commonly used model of PDAC. Patient tumour is implanted subcutaneously or orthotopically in severe combined immune deficiency (SCID) mice until the tumour has grown to a sufficient size to be sub-cultured in new mice. These models allow for tumours to have the original cell-to-cell interactions [103]. The original tumour microenvironment can also be recapitulated using orthotopic implants. A study by Garrido-Laguna et al. [104] showed that orthotopic implantation closely mirrors the results from the clinic. Orthotopically implanted tumours treated with gemcitabine had a similar response to that in patients, which was not observed in the subcutaneous implanted tumours.
PDX studies have been used for the identification of biomarkers of PDAC. Jimeno et al. [105] used 11 PDX tumour samples, with known gemcitabine sensitivity to identify biomarkers for gemcitabine response in patients. This group exposed fine-needle biopsy of the PDX tumour to gemcitabine or vehicle control for 6 h and compared gene expression of the treated and untreated samples using qRT-PCR 45-gene array. This assay identified that Polo-Like Kinase 1 (Plk1), a serine/threonine-protein kinase had differential expression of >50% in the sensitive samples compared to resistant tumours. To further validate this biomarker, the group performed siRNA knockdown and inhibition of the Plk1 pathway using a pathway modulator which resulted in a synergistic effect with gemcitabine in gemcitabine-resistant in vitro models. The study illustrates the ability to use PDX models to identify and validate a biomarker of PDAC.
There are many advantages to using PDX tumours for the study of pancreatic cancer. Tumours can be established in mice using a small amount of tumour; tumours retain the heterogeneity, as well as the genetics, and histological characteristics of the original tumour during passaging. PDX tumours also provide an unlimited source of tumour, which can be used for in vivo and ex vivo drug testing. Nevertheless, there are several disadvantages to the use of PDX models—they are expensive, time consuming, require the use of animals, and their use is subject to strict regulations [106]. PDX models take up to four months to develop tumours. Subcutaneously implanted tumours are not grown in the same microenvironment as PDAC tumours, and rarely form metastases [107]. As the tumour is grown, and sub-cultured in mice, the human stromal cells, such as fibroblasts and blood vessels are replaced by murine cells [108]. Finally, as SCID mice do not have an immune system, the PDX tumours cannot recapitulate the complex interactions between the PDAC tumour and the immune system, which is critical in resistance mechanisms of PDAC, and it also prevents the use of PDX models in testing of immune modulating drugs, which are increasingly being used in cancer treatment.
3.5. Genetically Engineered Mouse Models (GEMM)
Genetically engineered mouse models of PDAC can be used for both basic and translational cancer research. GEMM develop de novo tumours in an immune proficient environment, mimic the histopathological and molecular features of human tumours [109]. They also spontaneously develop metastatic disease [109]. With the use of CRISPR genome editing technology allowing for site directed double strand breaks resulting in gene knockouts and the introduction of defined mutations, the development of GEMM has become easier [110]. The KRASLSL.G12D/+; Pdx-1-Cre (KC) inducible knock-in GEMM, presents with slow disease progression, results in the development of Pancreatic Intraepithelial Neoplasia (PanIN). At a low frequency, these PanINs can also develop into locally invasive, and metastatic adenocarcinoma, allowing for the use of the KC model to study PanIN development and strategies to delay PDAC [111].
In order to overcome the challenges in studying immune-related drugs, GEMM contribute to immune research in PDAC. These models most include the most commonly mutated genes in PDAC, such as KRAS, TP53, SMAD4 and CDKN2A. The LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre (KPC) model is one of the most commonly used models for studying immunotherapy in PDAC. This model has the same features of the immune microenvironment as human PDAC, including the exclusion of effector T-cells [112]. The KPC model utilises a Cre-Lox technology, with the KRASG12D and TP53R172H mutations, with the Cre-recombinase activated by pancreas specific transcription factor PDX1. In this model, the new-born mouse has a normal pancreas, with PanIN development beginning at 8–10 weeks, with epithelial to mesenchymal markers such as decreased expression of E-cadherin, and increased expression of Zeb1 and Fsp1. At 16 weeks, the mice have developed locally invasive PDAC and the mice display cachexia, jaundice, weight loss malignant ascites and metastases. The KPC PDAC mouse model has been used in many pre-clinical studies, including Olive et al. [113] who studied the co-administration of gemcitabine and IPI-926, a drug which inhibits the Hedgehog signalling pathway to deplete tumour associated stroma, which resulted in an increased intertumoral concentration of gemcitabine. Frese et al. [114] used the model to study the effect of paclitaxel in combination with gemcitabine, and found increased intertumoral levels of gemcitabine due to the decreased levels of cytidine deaminase, which metabolises gemcitabine.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12051233