MHCs are the key components of the adaptive immune system that recognize foreign proteins. They are expressed on the surface of most nucleated non-immune and immune cells and present antigenic peptide fragments to either CD8
+ or CD4
+ T-cells for an adaptive immune response [
8,
9,
10]. The general structure of an MHC is composed of immunoglobulin-like anchoring peptides, which fixes the MHC to the exterior of cellular membrane and a peptide-binding region (PBR), which is responsible for antigen recognition and presentation to the T-cell receptors (TCR). MHC molecules are further subcategorized into class I and class II molecules [
8,
9,
10,
11,
12]. MHC class I molecules are heterodimeric molecules that are made up of two polypeptide chains: an α chain that is comprised of three domains (α1, α2, α3) and a smaller β
2-microglobulin chain [
13]. The α1 and α2 domains are key components of the PBR on MHC I and their inherent polymorphisms mitigate and influence antigenic peptides binding to the PBR for antigen presentation to CD8
+ CTLs [
9,
12,
13]. MHC class I molecules are consistently expressed on the surfaces of most nucleated cells except for sperm cells and select neuronal cells. MHC class II molecules are also heterodimeric molecules that are composed of an α chain and a β chain; however, they have two distinct domains referred to as α1, α2 and β1, β2 [
8,
12]. The PBR in MHC II is composed of α1 and β1 domains [
8,
10,
12]. MHC class II molecules are only expressed on antigen presenting cells (APCs) such as dendritic cells (DCs), macrophages, and B-cells, which specifically present to CD4
+ T cells for an immune response [
10,
12,
14].
2.1. Classification of Tumor Antigens
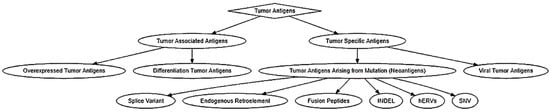
Tumor antigens were classified into two categories: tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs) (
Figure 1). TAAs are tumor antigens that are expressed in normal germline cells as well as tumor cells [
23]. Due to the wide expression profile of TAAs, they have been further subcategorized into differentiation tumor antigens and overexpressed tumor antigens [
23]. Overexpressed tumor antigens are a class of TAAs that could be found in normal tissues but are expressed at an elevated level in various cancerous tissues [
23,
24]. A well-known example is human epidermal growth factor receptor 2 (also known as HER2 or ERBB2). Differentiation tumor antigens are a class of TAAs that are limited in their expression to one tissue type and show lineage-specific expression. These antigens are expressed in tumors and normal cells that are derived from the same cellular origin [
24,
25]. Antigens derived from melanocyte differentiation proteins are typical differentiation antigens that are expressed in melanomas but can be expressed in normal skin melanocytes and retinal tissue [
25,
26]. Due to the “self” nature of these antigens, there will be a propensity for the development of autoimmune disorders, such as the occurrence of vitiligo after the chemoimmunotherapy of metastatic melanoma [
27]. More examples of TAAs and TSAs were summarized in the
supplementary tables.
Figure 1. Tumor antigen classifications.
TSAs are cancer antigens that are specifically expressed in malignant cancer cells and have a wide variety of expression among different cancer types. For example, the expression of cancer/testis antigens are restricted to the testis and ovary in normal tissues, but they could be found in a wide range of human tumors [
28,
29]. Neoantigens are tumor antigens that are only expressed in tumor cells and usually arise from non-synonymous single-nucleotide variant mutations (SNVs), but non-mutated neoantigens have also been identified [
8,
23,
30]. Tumor antigens arising from mutations are TSAs that are expressed ubiquitously across all cell types but are mutated when they are expressed in tumors. These mutations could arise from SNV, insertion and deletion (INDEL), gene fusion, splice variant, endogenous retroelement, and human endogenous retrovirus mutations (hERVs) which induce a change in the amino acid sequence of a protein [
24,
25,
31,
32,
33,
34]. These changes in the amino acid sequences further differentiate a protein from its normal expression profile in non-cancerous cells, subsequently allowing the generation of novel peptide epitopes that can then participate in restrictive presentation on MHC molecules for an adaptive immune response [
24,
25]. For example, the transcript for the BCR-ABL fusion protein is formed via reciprocal translocation between chromosomes 9 and 22 [
35]. The chromosomal abnormality was first identified in cases of chronic myeloid leukemia (CML) [
36]. BCR-ABL has been shown to be expressed in more than 95% of CML cases [
37], but it is also present in approximately 10% to 20% of adults and 2% to 5% of children with acute lymphoblastic leukemia (ALL) [
38,
39,
40], and in some cases of acute myeloid leukemia (AML) [
41,
42,
43], lymphomas [
44,
45,
46], and myelomas [
47,
48,
49]. Besides, K-ras protein is found to be highly mutated in cancers with the bulk of the mutations occurring at position 12 which is termed KRAS G12D, with the change from glycine (G) to aspartic acid (D) or valine (V) [
50].
Table 1 provides a summary of neoantigens and their HLA classes. Antigens derived from oncogenic virus comprise another relatively small but indispensable member of the TSA family [
24]. The best examples of viral tumor antigens are E6 and E7 oncogenic proteins derived from human papillomavirus 16 (HPV-16). The E6 oncogenic protein is capable of producing epitopes for restrictive presentation on the class-I HLA molecule HLA-B18 for MHC-I [
51]. The E7 oncogenic protein yielded an epitope capable of binding in a restrictive fashion on the class-I HLA molecules HLA-A2 and HLA-B18 for MHC-I and on the class-II HLA molecule HLA-DQ2 for MHC-II [
51,
52,
53].
Table 1. Tumor antigens arising from mutations.
2.2. Biochemical and Bioinformatic Approaches for the Identification of Tumor Antigens
2.2.1. Serological Analysis of Recombinant Tumor cDNA Libraries (SEREX)
SEREX is a technique developed by Sahin, Pfreundschuh et al., whereby one can identify novel tumor antigens through the sampling of mRNA from fresh tumor specimens, instead of in vitro cancer cell lines [
106]. This is because in vitro cancer cell lines can be subject to either a loss or unwanted generation of cancer antigens, due to mutations that might arise during the continuance of the cell culture. The mRNA extracted from the fresh tumor specimen is used to build a cDNA library and subsequently cloned into a λ phage expression vector. This λ phage expression vector is transfected into
Escherichia coli for the recombinant expression of potential cancer antigens in the cDNA library. Recombinant proteins are collected and transferred onto a nitrocellulose membrane, blocked, and exposed to autologous diluted serum (1:100 or 1:1000) from the same patient that tumor specimens are taken from. The serum is diluted to ensure that only high-titer IgG antibodies react with the recombinant proteins on the nitrocellulose membrane. Subsequently, a secondary immunoscreening is performed with anti-human IgG for the purification and identification of positive clones while eliminating the false positives that can arise from residual recombinant immunoglobulin (IgG) expression, due to the B-cells and plasma cells that are present in the tumor specimen being sampled. Finally, positive clones are subcloned for isolation of that specific antigenic cDNA fragment and that cDNA fragment is sequenced to determine its nucleotide sequence [
107,
108,
109]. One of the major drawbacks of SEREX is that the bacteria is incapable of expressing low abundant TAAs and their tumor-specific post-translational modification [
110,
111]. SEREX-defined antigens are usually weakly immunogenic due to the lack of mutations or structural aberrance [
112]. SEREX has also been criticized for its demanding protocol and poor reproducibility [
111].
2.2.2. Computational Prediction Methods for Cancer Antigens
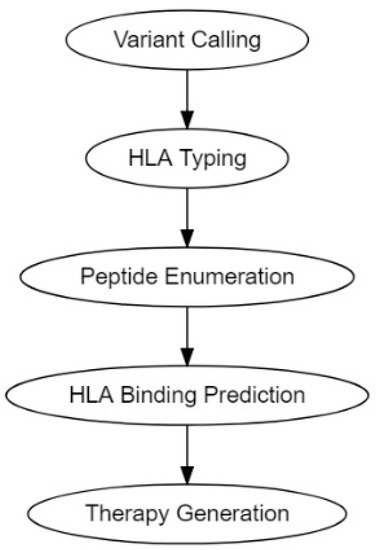
The traditional pipeline for computational tumor-specific antigen prediction segments itself into five distinct steps: variant calling, HLA typing, peptide enumeration, HLA binding prediction, and finally therapy generation (
Figure 2) [
30,
113]. Variant calling involves predicting potential cancer antigens through methods that use data from high-throughput genetic sequencing (RNA-Seq or DNA-Seq). This genetic data is processed through algorithms best suited to predict the potential antigenicity of a TSA, depending on its mutational origin: SNV, INDEL, frameshifts, fusion proteins, endogenous retroelement, or hERVs [
30,
113,
114]. HLA typing is performed to determine HLA allele frequencies [
30,
113,
115]. Peptide enumeration is done to determine the peptide sequences of the potential antigenic mutants and to sort them from incompatible antigenic sequences that arise from non-sense mutations and other non-functional genetic aberrations [
30,
113,
116]. HLA binding prediction is enacted to determine the binding of affinity of the antigenic peptide to the corresponding HLA molecule. This HLA-to-peptide affinity quantification is usually expressed as either a ranked percentile or a K
D ≤ 500 nM (standard cutoff for detection) [
80,
117]. Finally, the genetic information gathered was utilized to make vaccine and cellular therapeutics such as DNA, RNA, peptide, and autologous DC or T-cell vaccines.
Figure 2. Tumor antigen computational pipelines.
Computational prediction pipelines such as Epidisco (ver.1.0, lex Rubinsteyn, et al.), Antigen.garnish (ver.2.3.1, Andrew J Rech, Lee Richman), pVACtools (ver.3.0.2, Jasreet Hundal, et al.), Neopepsee (ver.3.0.1, Sora Kim), MuPeXI (ver.1.2.0, Anne-Mette Bjerregaard, Aron C. Eklund), TSNAD (ver.2.0.1, Zhan Zhou et al.), NeoepitopePred (ver.1.0, Jinghui Zhang et al.), and INTEGRATE-Neo (ver.1.2.1, Jin Zhang, et al.) condense these steps (variant calling, HLA typing, peptide enumeration, HLA binding prediction) into succinct computational workflows [
118,
119,
120,
121,
122,
123,
124,
125]. Epidisco is very versatile in the variety of TSAs it can predict. It specializes in identifying potential TSA neoantigens of SNV, INDEL, splice variant, and gene fusion mutational origins that bind MHC class I molecules [
123]. Antigen.garnish and pVACtools also have extensive predictive abilities, with both being able to predict TSA neoantigens of SNV, INDEL, and gene fusion mutational origins [
120,
122]. However, unlike Epidisco and pVACtools, which only allow for MHC-I antigen prediction, Antigen.garnish can do both MHC class I and class II binding predictions for the neoantigens it predicts [
120,
122,
123]. MuPeXI, TSNAD, and Neopepsee are all used to predict TSAs that arise from SNV and INDEL mutational origins [
118,
121,
125]. Even though MuPeXI, TSNAD, and Neopepsee all work to call the same mutational variants (SNV and INDEL); Neopepsee can only make binding predictions for MHC-I whereas MuPeXI and TSNAD can make binding predictions for both MHC-I and MHC-II [
118,
121,
125]. NeoepitopePred works to predict SNV, and gene fusion TSA mutational variants and can make MHC-peptide binding predictions for MHC-I [
119]. INTEGRATE-Neo is a very specialized workflow in that it only predicts TSA mutational variants of gene fusion origins for binding to MHC class I molecules [
124]. Although there are currently no programing pipe lines in place that specialize in the MHC binding prediction for TSAs originating from retroelements or human endogenous retroviruses (hERVs), RepeatMasker and hervQuant are variant calling tools (not complete computational pipelines) that can be used to identify potential TSAs originating from retroelements and hERVs respectively [
126,
127].
2.3. Delivery of Neoantigens
Neoantigens can be delivered in the form of synthetic long peptides or neoepitope-encoding mRNA or DNA [
5]. Direct injection of soluble subunit antigens and immune adjuvants can only induce modest immune responses due to their uncontrolled systemic distribution and poor targeting and accumulation in lymphoid organs. DNA requires electroporation-facilitated delivery and extra processing before presentation by DCs [
5,
128]. mRNA needs delivery platforms to facilitate the intracellular delivery and protect it from ribonuclease degradation [
129,
130]. Therefore, neoantigen vaccines formulated with novel technologies and biomaterials, such as lipids and biodegradable polymers, are pursued to improve the safety and efficiency of neoantigen delivery. Current progress in the delivery of neoantigen vaccines has been discussed in other reviews [
131,
132].
3. Vaccine Adjuvants
Adjuvants are known as a variety of substances used in combination with a specific antigen that produce stronger immunity than the antigen alone [
133]. Incorporating an adjuvant in a vaccine does not only strengthen the adaptive response to antigens but also enables a comparable response with a lower dose of antigens or less frequent vaccinations relative to unadjuvanted vaccines [
134,
135]. As such, adjuvants have become essential components of many successful vaccines and those still in clinical trials. For example, the adjuvant system 04 (AS04), consisting of aluminum salt particles loaded with MPL (3-
O-desacyl-4′-monophosphoryl lipid A), has been used in two licensed vaccines, Cervarix
TM against human papillomavirus (HPV) and Fendrix
TM against hepatitis B virus [
136,
137]. Poly ICLC is a derivative of toll-like receptor (TLR) 3 agonist polyriboinosinic-polyribocytidylic acid (poly(I:C)) stabilized with carboxymethylcellulose and poly-
L-lysine [
138], which has been widely utilized as an adjuvant in therapeutic vaccines against different cancers in more than 50 clinical trials [
139].
From a mechanistic view, an immune response starts with sampling and presentation of antigens by APCs. Evidence has demonstrated that adjuvants can activate APCs, facilitate antigen uptake and cross-presentation between APCs and T-cells, and stimulate the production of immunoregulatory molecules [
147,
148]. In addition, the importance of adjuvants is underlined when they are exploited to direct the desired types of immune response (e.g., type-1 immunity versus type-2 immunity, CD8
+ versus CD4
+ T-cells) and promote the generation of immunological memory [
135]. Based on their modes of action, adjuvants can be grouped into two main categories, delivery systems and immune potentiators [
141]. Delivery systems act as carriers or depots where antigens and other vaccine components can stay and maintain their stability; in the meantime, they create local proinflammatory responses and recruit APCs. Immune potentiators can activate innate immune cells directly or through PRRs, such as TLRs, nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs) [
135]. The interactions between PRRs and pathogen-associated molecular patterns (PAMPs) activate innate immune cells to produce chemokines and cytokines [
140]. Once activated, APCs will present antigens to T-cells via MHC and release costimulatory molecules to prime naïve T-cells, bridging the fast-acting innate response with antigen-specific adaptive response.