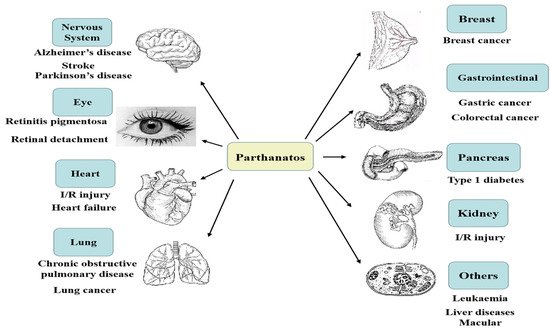
Differential evolution of apoptosis, programmed necrosis, and autophagy, parthanatos is a form of cell death mediated by poly(ADP-ribose) polymerase 1 (PARP1), which is caused by DNA damage. PARP1 hyper-activation stimulates apoptosis-inducing factor (AIF) nucleus translocation, and accelerates nicotinamide adenine dinucleotide (NAD+) and adenosine triphosphate (ATP) depletion, leading to DNA fragmentation. The mechanisms of parthanatos mainly include DNA damage, PARP1 hyper-activation, PAR accumulation, NAD+ and ATP depletion, and AIF nucleus translocation. Parthanatos, a kind of new programmed death mode, has been put forward by Professors Ted and Valina Dawson to indicate a caspase-independent cell death subroutine that critically relies on the hyper-activation of poly(ADP-ribose) polymerase 1 (PARP1).
- parthanatos
- hallmarks
- molecular mechanisms
1. Introduction
| Features | Parthanatos | Apoptosis | Necroptosis | Autophagy |
|---|---|---|---|---|
| Morphological features | Dissipation of the inner transmembrane potential, nuclear and chromatin condensation | Cellular and nuclear volume reduction, chromatin agglutination, nuclear fragmentation, formation of apoptotic bodies and cytoskeletal disintegration, no significant changes in mitochondrial structure | Plasma membrane breakdown, generalized swelling of the cytoplasm and organelles, moderate chromatin condensation, spillage of cellular constituents into the microenvironment | Formation of double-membraned autolysosomes, including macroautophagy, microautophagy and chaperone-mediated autophagy |
| Biochemical features | DNA injury, energy depletion and PAR accumulation | DNA fragmentation | Drop in ATP levels | Increased lysosomal activity |
| Regulatory pathways | PARP1/AIF signaling pathway | Death receptor pathway, mitochondrion pathway and endoplasmic reticulum pathway; caspase, P53, Bcl-2-mediated signaling pathway | Tumor necrosis factor type 1 (TNF-R1) and Receptor-interacting protein 1 (RIP1)/RIP3-mixed-lineage kinase domain-like (MLKL) related signaling pathways; protein kinase C (PKC)-mitogen-activated protein kinase (MAPK)-activatorprotein1 (AP1) related signaling pathway; ROS-related metabolic regulation pathway | Molecular target of rapamycin (mTOR), Beclin-1, P53 signaling pathway |
| Key genetic inhibition or inhibition by protein overexpression | PARP1 knockout, AIF down-regulation (e.g., in Harlequin mouse) |
Bcl-2 overexpression, Inhibition of caspases (3, 8, and 9), Inhibition of PP2Ad, CrmA expression |
Inhibition of RIP1 or RIP3 | Inhibition of Activating molecule in BECN1-regulated autophagy protein 1 (AMBRA1), Recombinant human autophagy related 5/7/12 (ATG5/7/12), or Recombinant Beclin 1 (BECN1) |
| Examples of trigger factors and/or conditions | Excitotoxicity Ischemia Stroke Reactive oxygen/nitrogen species |
Death receptor signaling Dependence receptor signaling DNA damage Trophic factor withdrawal Viral infections |
Excitotoxicity Ischemia Stroke Reactive oxygen/nitrogen species |
Amino acid starvation Serum starvation Protein aggregates |
2. Hallmarks of Parthanatos
2.1. Morphological Features
2.2. Biochemical Features
2.2.1. DNA Injury
2.2.2. NAD+ Depletion
2.2.3. Poly(ADP-Ribose) (PAR) Accumulation
2.3. Genetic Features
2.4. Immune Features
3. Molecular Mechanisms of Parthanatos
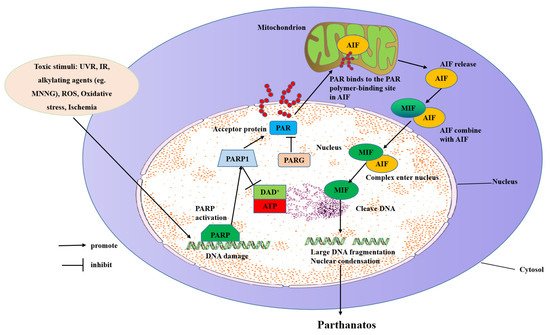
3.1. Inducing Parthanatos by Injuring DNA
3.2. Inducing Parthanatos by Hyper-Activating PARP1
3.3. Inducing Parthanatos by Binding of PAR
Nuclear DNA damage stimulation triggers the synthesis of poly ribose (ADP-ribose), next to the distribution of the PAR polymer to the cytoplasm and mitochondria, and then the release of AIF [13]. PAR inhibits the glycolysis process. ARH3 hydrolyzes protein-free PAR in the nucleus and cytoplasm, inhibits PAR transfer to the cytoplasm, and thus prevents parthanatos [62]. ARH3 has been shown to catalyze PAR-degradation in vitro [63][64]. PARG, the most well-characterized enzyme in humans for PAR hydrolysis, utilizes a macrodomain fold to bind ADP-ribosylation and specifically cleaves the ribose-ribose bonds between the subunits of the PAR chains. PARG needs the cooperation of PARP to repair DNA. Once the strand of PARP and DNA breaks, an enzyme is activated, causing PARP to shuttle and the chromatin to open. PARG enters the nucleus and moves to the PARP substrate to repair the broken DNA strand. With the increase in PARG and the decrease in PAR, chromatin recovers its original structure. PARG has been shown to play a vital role in various diseases [65].
3.4. Inducing Parthanatos by Depleting ATP and NAD+
NAD+ is a factor of ATP generation and its resynthesis needs many molecules of ATP, which its consumption results in ATP depletion and cellular energy downturn, which leads to parthanatos [7][28]. The inhibition of PARP1 by pharmacological inhibitors or genetic deletion could recover NAD+ levels [28]. Neurons that are stimulated with toxic show protection and accompanying energy conservation with PARP inhibitors. Besides, PARP1 KO mice of transient cerebral ischemia-induced damage present preserved NAD+ levels [29][66], supporting the suicide hypothesis of PARP1 activation causing a block in glycolysis. Besides, adding NAD+ to cells or over-expression of NAD+ avoids PARP1-dependent parthanatos, which suggests that the decreased NAD+ relevant to PARP1 hyper-activation causes cell demise [28].
3.5. Inducing Parthanatos by Releasing and Translocating AIF from Mitochondrial to the Nucleus
Mitochondria is a crucial element in the regulation of parthanatos. Its inter-membrane gap contains a number of proteins released through the outer membrane for taking part in parthanatos degradation. Mitochondria maintain the physiological integrity of cells by energy generation. However, once a cell is stimulated, the mitochondria become permeabilized, and AIF is released to irritate parthanatos. The change of outer membrane permeabilization (OMP) and permeability transition pore (PTP) will lead to the release of pro-apoptotic factors [77]. Bax, one of the pro-apoptotic members, is downstream of PARP1 and induces OMP needed for AIF release. Besides OMP, AIF cleavage by calpains requires AIF to be entirely released from the mitochondria [9].
References
- Aki, T.; Funakoshi, T.; Uemura, K. Regulated necrosis and its implications in toxicology. Toxicology 2015, 333, 118–126.
- Galluzzi, L.; Kepp, O.; Krautwald, S.; Kroemer, G.; Linkermann, A. Molecular mechanisms of regulated necrosis. Semin. Cell Dev. Biol. 2014, 35, 24–32.
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364.
- Liu, S.; Luo, W.; Wang, Y. Emerging role of PARP1 and PARthanatos in ischemic stroke. J. Neurochem. 2021, 160, 74–87.
- Kong, D.; Zhu, J.; Liu, Q.; Jiang, Y.; Xu, L.; Luo, N.; Zhao, Z.; Zhai, Q.; Zhang, H.; Zhu, M.; et al. Mesenchymal stem cells protect neurons against hypoxic-ischemic injury via inhibiting parthanatos, necroptosis, and apoptosis, but not autophagy. Cell. Mol. Neurobiol. 2017, 37, 303–313.
- Power, M.; Das, S.; Schutze, K.; Marigo, V.; Ekstrom, P.; Paquet-Durand, F. Cellular mechanisms of hereditary photoreceptor degeneration—Focus on cGMP. Prog. Retin. Eye Res. 2020, 74, 100772.
- Zhou, Y.; Liu, L.; Tao, S.; Yao, Y.; Wang, Y.; Wei, Q.; Shao, A.; Deng, Y. Parthanatos and its associated components: Promising therapeutic targets for cancer. Pharmacol. Res. 2021, 163, 105299.
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)—Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front. Mol. Biosci. 2020, 28, 191.
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Parthanatos, a messenger of death. Front. Biosci (Landmark Ed.) 2009, 14, 1116–1128.
- Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Mitochondrial and nuclear cross talk in cell death: Parthanatos. Ann. N. Y. Acad. Sci. 2008, 1147, 233–241.
- Wang, H.; Yu, S.W.; Koh, D.W.; Lew, J.; Coombs, C.; Bowers, W.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J. Neurosci. 2004, 24, 10963–10973.
- Aredia, F.; Scovassi, A.I. Involvement of PARPs in cell death. Front. Biosci (Elite Ed.) 2014, 6, 308–317.
- Virag, L.; Robaszkiewicz, A.; Rodriguez-Vargas, J.M.; Oliver, F.J. Poly(ADP-ribose) signaling in cell death. Mol. Aspects Med. 2013, 34, 1153–1167.
- Kim, J.H.; Kim, J.; Roh, J.; Park, C.S.; Seoh, J.Y.; Hwang, E.S. Reactive oxygen species-induced parthanatos of immunocytes by human cytomegalovirus-associated substance. Microbiol. Immunol. 2018, 62, 229–242.
- Wang, Y.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) signals to mitochondrial AIF: A key event in parthanatos. Exp. Neurol. 2009, 218, 193–202.
- Santivasi, W.L.; Xia, F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid. Redox Signal. 2014, 21, 251–259.
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120.
- Andrabi, S.A.; Umanah, G.K.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagne, J.P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214.
- Bedard, L.L.; Massey, T.E. Aflatoxin B1-induced DNA damage and its repair. Cancer Lett. 2006, 241, 174–183.
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084.
- Salehi, F.; Behboudi, H.; Kavoosi, G.; Ardestani, S.K. Oxidative DNA damage induced by ROS-modulating agents with the ability to target DNA: A comparison of the biological characteristics of citrus pectin and apple pectin. Sci. Rep. 2018, 8, 13902.
- Wang, Y.; Luo, W.; Wang, Y. PARP1 and its associated nucleases in DNA damage response. DNA Repair 2019, 81, 102651.
- Stringari, C.; Edwards, R.A.; Pate, K.T.; Waterman, M.L.; Donovan, P.J.; Gratton, E. Metabolic trajectory of cellular differentiation in small intestine by Phasor Fluorescence Lifetime Microscopy of NADH. Sci. Rep. 2012, 2, 568.
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007, 32, 12–19.
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31.
- Berger, N.A.; Berger, S.J. Metabolic consequences of DNA damage: The role of poly (ADP-ribose) polymerase as mediator of the suicide response. Basic Life Sci. 1986, 38, 357–363.
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982.
- Chiarugi, A. Intrinsic mechanisms of poly(ADP-ribose) neurotoxicity: Three hypotheses. Neurotoxicology 2005, 26, 847–855.
- Yu, S.W.; Wang, H.; Dawson, T.M.; Dawson, V.L. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol. Dis. 2003, 14, 303–317.
- Hegedus, C.; Boros, G.; Fidrus, E.; Kis, G.N.; Antal, M.; Juhasz, T.; Janka, E.A.; Janko, L.; Paragh, G.; Emri, G.; et al. PARP1 Inhibition Augments UVB-Mediated Mitochondrial Changes-Implications for UV-Induced DNA Repair and Photocarcinogenesis. Cancers 2019, 12, 5.
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43.
- Dawson, V.L.; Dawson, T.M. Deadly conversations: Nuclear-mitochondrial cross-talk. J. Bioenerg. Biomembr. 2004, 36, 287–294.
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016.
- Krietsch, J.; Caron, M.C.; Gagné, J.P.; Ethier, C.; Vignard, J.; Vincent, M.; Rouleau, M.; Hendzel, M.J.; Poirier, G.G.; Masson, J.Y. PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012, 40, 10287–10301.
- Andrabi, S.A.; Kim, N.S.; Yu, S.W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313.
- Gagné, J.P.; Isabelle, M.; Lo, K.S.; Bourassa, S.; Hendzel, M.J.; Dawson, V.L.; Dawson, T.M.; Poirier, G.G. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008, 36, 6959–6976.
- Wang, Y.; Kim, N.S.; Haince, J.F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011, 4, ra20.
- Yu, S.W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319.
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263.
- Rongvaux, A.; Galli, M.; Denanglaire, S.; Van Gool, F.; Dreze, P.L.; Szpirer, C.; Bureau, F.; Andris, F.; Leo, O. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J. Immunol. 2008, 181, 4685–4695.
- Culmsee, C.; Zhu, C.; Landshamer, S.; Becattini, B.; Wagner, E.; Pellecchia, M.; Blomgren, K.; Plesnila, N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J. Neurosci. 2005, 25, 10262–10272.
- Amé, J.C.; Fouquerel, E.; Gauthier, L.R.; Biard, D.; Boussin, F.D.; Dantzer, F.; de Murcia, G.; Schreiber, V. Radiation-induced mitotic catastrophe in PARG-deficient cells. J. Cell Sci. 2009, 122, 1990–2002.
- Shirai, H.; Poetsch, A.R.; Gunji, A.; Maeda, D.; Fujimori, H.; Fujihara, H.; Yoshida, T.; Ogino, H.; Masutani, M. PARG dysfunction enhances DNA double strand break formation in S-phase after alkylation DNA damage and augments different cell death pathways. Cell Death Dis. 2013, 4, e656.
- Palazzo, L.; Leidecker, O.; Prokhorova, E.; Dauben, H.; Matic, I.; Ahel, I. Serine is the major residue for ADP-ribosylation upon DNA damage. eLife 2018, 26, e34334.
- Jagtap, P.; Szabo, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440.
- Hansson, M.; Asea, A.; Ersson, U.; Hermodsson, S.; Hellstrand, K. Induction of apoptosis in NK cells by monocyte-derived reactive oxygen metabolites. J. Immunol. 1996, 156, 42–47.
- Aurelius, J.; Thoren, F.B.; Akhiani, A.A.; Brune, M.; Palmqvist, L.; Hansson, M.; Hellstrand, K.; Martner, A. Monocytic AML cells inactivate antileukemic lymphocytes: Role of NADPH oxidase/gp91(phox) expression and the PARP1/PAR pathway of apoptosis. Blood 2012, 119, 5832–5837.
- Aurelius, J.; Martner, A.; Brune, M.; Palmqvist, L.; Hansson, M.; Hellstrand, K.; Thoren, F.B. Remission maintenance in acute myeloid leukemia: Impact of functional histamine H2 receptors expressed by leukemic cells. Haematologica 2012, 97, 1904–1908.
- Akhiani, A.A.; Werlenius, O.; Aurelius, J.; Movitz, C.; Martner, A.; Hellstrand, K.; Thoren, F.B. Role of the ERK pathway for oxidant-induced parthanatos in human lymphocytes. PLoS ONE 2014, 9, e89646.
- Martinez-Morcillo, F.J.; Canton-Sandoval, J.; Martinez-Menchon, T.; Corbalan-Velez, R.; Mesa-Del-Castillo, P.; Perez-Oliva, A.B.; Garcia-Moreno, D.; Mulero, V. Non-canonical roles of NAMPT and PARP in inflammation. Dev. Comp. Immunol. 2021, 115, 103881.
- Wang, J.Q.; Tang, Y.; Li, Q.S.; Xiao, M.; Li, M.; Sheng, Y.T.; Yang, Y.; Wang, Y.L. PARG regulates the proliferation and differentiation of DCs and T cells via PARP/NF-κB in tumour metastases of colon carcinoma. Oncol. Rep. 2019, 41, 2657–2666.
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagenes. 2017, 58, 235–263.
- Gravells, P.; Grant, E.; Smith, K.M.; James, D.I.; Bryant, H.E. Specific killing of DNA damage-response deficient cells with inhibitors of poly(ADP-ribose) glycohydrolase. DNA Repair 2017, 52, 81–91.
- Prokhorova, E.; Zobel, F.; Smith, R.; Zentout, S.; Gibbs-Seymour, I.; Schützenhofer, K.; Peters, A.; Groslambert, J.; Zorzini, V.; Agnew, T.; et al. Serine-linked PARP1 auto-modification controls PARP inhibitor response. Nat. Commun. 2021, 12, 4055.
- Smith, S. The world according to PARP. Trends Biochem. Sci. 2001, 26, 174–179.
- Kim, M.Y.; Zhang, T.; Kraus, W.L. Poly(ADP-ribosyl)ation by PARP1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005, 19, 1951–1967.
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268.
- Keuss, M.J.; Hjerpe, R.; Hsia, O.; Gourlay, R.; Burchmore, R.; Trost, M.; Kurz, T. Unanchored tri-NEDD8 inhibits PARP1 to protect from oxidative stress-induced cell death. EMBO J. 2019, 38, 100024.
- Shall, S.; de Murcia, G. Poly(ADP-ribose) polymerase-1: What have we learned from the deficient mouse model? Mutat. Res. 2000, 460, 1–15.
- Martínez-Morcillo, F.J.; Cantón-Sandoval, J.; Martínez-Navarro, F.J.; Cabas, I.; Martínez-Vicente, I.; Armistead, J.; Hatzold, J.; López-Muñoz, A.; Martínez-Menchón, T.; Corbalán-Vélez, R.; et al. NAMPT-derived NAD+ fuels PARP1 to promote skin inflammation through parthanatos cell death. PLoS Biol. 2021, 19, e3001455.
- Thapa, K.; Khan, H.; Sharma, U.; Grewal, A.K.; Singh, T.G. Poly (ADP-ribose) polymerase-1 as a promising drug target for neurodegenerative diseases. Life Sci. 2021, 267, 118975.
- Mashimo, M.; Moss, J. ADP-Ribosyl-Acceptor Hydrolase Activities Catalyzed by the ARH Family of Proteins. Methods Mol. Biol. 2018, 1813, 187–204.
- Niere, M.; Mashimo, M.; Agledal, L.; Dölle, C.; Kasamatsu, A.; Kato, J.; Moss, J.; Ziegler, M. ADP-ribosylhydrolase 3 (ARH3), not poly(ADP-ribose) glycohydrolase (PARG) isoforms, is responsible for degradation of mitochondrial matrix-associated poly(ADP-ribose). J. Biol. Chem. 2012, 287, 16088–16102.
- Munnur, D.; Ahel, I. Reversible mono-ADP-ribosylation of DNA breaks. FEBS J. 2017, 284, 4002–4016.
- Fauzee, N.J.; Pan, J.; Wang, Y.L. PARP and PARG inhibitors—New therapeutic targets in cancer treatment. Pathol. Oncol. Res. 2010, 16, 469–478.
- Sheline, C.T.; Wei, L. Free radical-mediated neurotoxicity may be caused by inhibition of mitochondrial dehydrogenases in vitro and in vivo. Neuroscience 2006, 140, 235–246.
- Alano, C.C.; Tran, A.; Tao, R.; Ying, W.; Karliner, J.S.; Swanson, R.A. Differences among cell types in NAD(+) compartmentalization: A comparison of neurons, astrocytes, and cardiac myocytes. J. Neurosci. Res. 2007, 85, 3378–3385.
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107.
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santos-Argumedo, L.; Parkhouse, R.M.; Walseth, T.F.; Lee, H.C. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059.
- Paschen, W.; Olah, L.; Mies, G. Effect of transient focal ischemia of mouse brain on energy state and NAD levels: No evidence that NAD depletion plays a major role in secondary disturbances of energy metabolism. J. Neurochem. 2000, 75, 1675–1680.
- Goto, S.; Xue, R.; Sugo, N.; Sawada, M.; Blizzard, K.K.; Poitras, M.F.; Johns, D.C.; Dawson, T.M.; Dawson, V.L.; Crain, B.J.; et al. Poly(ADP-ribose) polymerase impairs early and long-term experimental stroke recovery. Stroke 2002, 33, 1101–1106.
- Moubarak, R.S.; Yuste, V.J.; Artus, C.; Bouharrour, A.; Greer, P.A.; Menissier-de Murcia, J.; Susin, S.A. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol. Cell. Biol. 2007, 27, 4844–4862.
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528.
- Kim, H.; Jacobson, E.L.; Jacobson, M.K. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science 1993, 261, 1330–1333.
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223.
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528.
- Beal, M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005, 58, 495–505.
- Klein, J.A.; Longo-Guess, C.M.; Rossmann, M.P.; Seburn, K.L.; Hurd, R.E.; Frankel, W.N.; Bronson, R.T.; Ackerman, S.L. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 2002, 419, 367–374.
- Wang, H.; Shimoji, M.; Yu, S.W.; Dawson, T.M.; Dawson, V.L. Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 132–139.
- Ye, H.; Cande, C.; Stephanou, N.C.; Jiang, S.; Gurbuxani, S.; Larochette, N.; Daugas, E.; Garrido, C.; Kroemer, G.; Wu, H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat. Struct. Biol. 2002, 9, 680–684.
- Mate, M.J.; Ortiz-Lombardia, M.; Boitel, B.; Haouz, A.; Tello, D.; Susin, S.A.; Penninger, J.; Kroemer, G.; Alzari, P.M. The crystal structure of the mouse apoptosis-inducing factor AIF. Nat. Struct. Biol. 2002, 9, 442–446.
- Boehler, C.; Gauthier, L.R.; Mortusewicz, O.; Biard, D.S.; Saliou, J.M.; Bresson, A.; Sanglier-Cianferani, S.; Smith, S.; Schreiber, V.; Boussin, F.; et al. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. USA 2011, 108, 2783–2788.
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978.
- Brosey, C.A.; Ho, C.; Long, W.Z.; Singh, S.; Burnett, K.; Hura, G.L.; Nix, J.C.; Bowman, G.R.; Ellenberger, T.; Tainer, J.A. Defining NADH-Driven Allostery Regulating Apoptosis-Inducing Factor. Structure 2016, 24, 2067–2079.