Microtubule poisons, as is the case with other antitumor drugs, routinely promote autophagy in tumor cells. However, the nature and function of the autophagy, in terms of whether it is cytoprotective, cytotoxic or nonprotective, cannot be predicted; this likely depends on both the type of drug studied as well as the tumor cell under investigation. The microtubule poisons continue to play a central role in the clinical treatment of both solid tumors or hematologic malignancies. However, tumor cells can develop resistance by a number of mechanisms such as altered microtubule binding and efflux via the multidrug resistance pump family of transporters.
- autophagy
- microtubule poison
- cytoprotective
- cytotoxic
1. Introduction
2. Direct Involvement of Microtubules in Autophagy
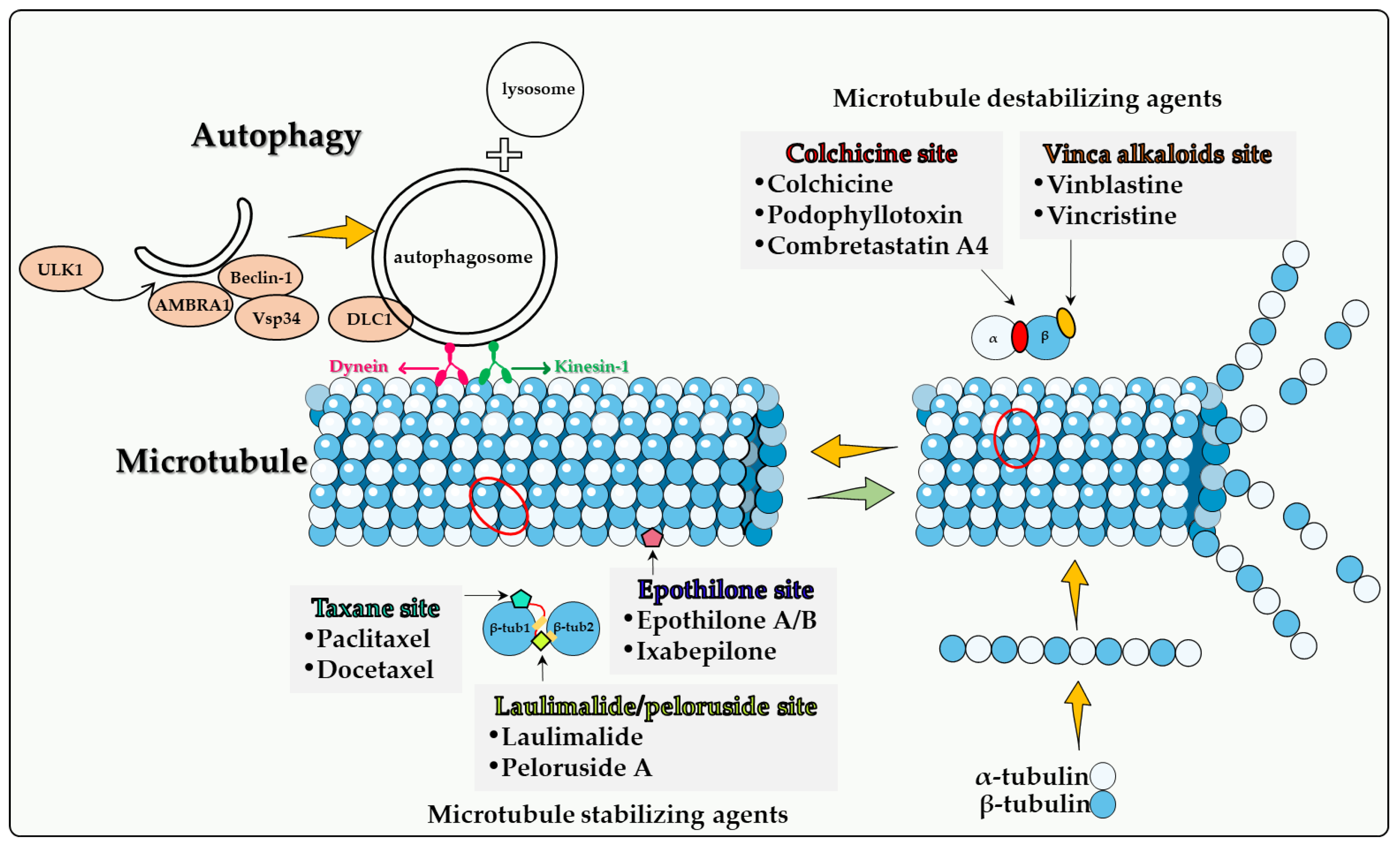
2.1. Colchicine Site
2.2. Vinca Alkaloid Site
2.3. Taxane Site
2.4. Epothilone Site
2.5. Laulimalide/Peloruside Site
3. Summary
Although many new cancer treatments have been developed in recent years, such as targeted therapy and immunotherapy, microtubule poisons remain a critical class of first-line chemotherapeutic agents. However, the relationship(s) between microtubule poisons and autophagy are exceedingly complex. This is likely due to a number of factors including that (a) microtubules play a direct role in the autophagic process; (b) the different microtubule poisons do not all act at the identical microtubule binding sites, as indicated in Figure 1; (c) autophagy cannot have only one (cytoprotective) function, but (at least) three others, termed cytotoxic, cytostatic and nonprotective. Although the cytoprotective function is often clearly induced, both the cytotoxic and nonprotective function have also been identified in response to these agents. This may be dependent upon the chemical structure of the drug as well as the experimental cell line being utilized. final and critical issue is that the literature evaluating the role of autophagy in response to microtubule poisons in tumor cells has generally relied on pharmacologic autophagy inhibitors such as CQ (or HCQ) and 3-MA drugs, which are not exclusively autophagy inhibitors [60]. In the absence of studies utilizing genetic autophagy inhibition, inferences related to the nature of the autophagy are not sufficiently well supported to be conclusive.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10071632
References
- Caplow, M. Microtubule dynamics. Curr. Opin. Cell Biol. 1992, 4, 58–65.
- Carlier, M.F.; Hill, T.L.; Chen, Y. Interference of GTP hydrolysis in the mechanism of microtubule assembly: An experimental study. Proc. Natl. Acad. Sci. USA 1984, 81, 771–775.
- Jánosi, I.M.; Chrétien, D.; Flyvbjerg, H. Structural microtubule cap: Stability, catastrophe, rescue, and third state. Biophys. J. 2002, 83, 1317–1330.
- Mitchison, T.J.; Kirschner, M.W. Dynamic instability of microtubule growth. Nature 1984, 312, 237–242.
- McKean, P.G.; Vaughan, S.; Gull, K. The extended tubulin superfamily. J. Cell. Sci. 2001, 114, 2723–2733.
- Goodson, H.V.; Jonasson, E.M. Microtubules and microtubule-associated proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608.
- Cutts, J.H.; Beer, C.T.; Noble, R.L. Biological properties of Vincaleukoblastine, an alkaloid in Vinca rosea Linn, with reference to its antitumor action. Cancer Res. 1960, 20, 1023–1031.
- Schneider, F.; Pan, L.; Ottenbruch, M.; List, T.; Gaich, T. The chemistry of nonclassical taxane diterpene. Acc. Chem. Res. 2021, 54, 2347–2360.
- Yang, C.H.; Horwitz, S.B. Taxol(R): The first microtubule stabilizing agent. Int. J. Mol. Sci. 2017, 18, 1733.
- Huitorel, P. From cilia and flagella to intracellular motility and back again: A review of a few aspects of microtubule-based motility. Biol. Cell 1988, 63, 249–258.
- Forth, S.; Kapoor, T.M. The mechanics of microtubule networks in cell division. J. Cell Biol. 2017, 216, 1525–1531.
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728.
- Jahreiss, L.; Menzies, F.M.; Rubinsztein, D.C. The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008, 9, 574–587.
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent Movement of Autophagosomes Mediates Efficient Encounters with Lysosomes. Cell Struct. Funct. 2008, 33, 109–122.
- Geeraert, C.; Ratier, A.; Pfisterer, S.G.; Perdiz, D.; Cantaloube, I.; Rouault, A.; Pattingre, S.; Proikas-Cezanne, T.; Codogno, P.; Poüs, C. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 2010, 285, 24184–24194.
- Luo, S.; Garcia-Arencibia, M.; Zhao, R.; Puri, C.; Toh, P.P.; Sadiq, O.; Rubinsztein, D.C. Bim Inhibits autophagy by recruiting beclin 1 to microtubules. Mol. Cell 2012, 47, 359–370.
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunra, L. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168.
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125.
- Kast, D.J.; Dominguez, R. The cytoskeleton–autophagy connection. Curr. Biol. 2017, 27, R318–R326.
- Fass, E.; Shvets, E.; Degani, I.; Hirschberg, K.; Elazar, Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 2006, 281, 36303–36316.
- Köchl, R.; Hu, X.W.; Chan, E.Y.W.; Tooze, S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 129–145.
- Nowosad, A.; Creff, J.; Jeannot, P.; Culerrier, R.; Codogno, P.; Manenti, S.; Nguyen, L.; Besson, A. p27 controls autophagic vesicle trafficking in glucose-deprived cells via the regulation of ATAT1-mediated microtubule acetylation. Cell Death Dis. 2021, 12, 481.
- Bhattacharyya, B.; Panda, D.; Gupta, S.; Banerjee, M. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med. Res. Rev. 2008, 28, 155–183.
- Sivakumar, G. Colchicine semisynthetics: Chemotherapeutics for cancer? Curr. Med. Chem. 2013, 20, 892–898.
- Arthur, C.R.; Gupton, J.T.; Kellogg, G.; Yeudall, W.A.; Cabot, M.C.; Newsham, I.F.; Gewirtz, D.A. Autophagic cell death, polyploidy and senescence induced in breast tumor cells by the substituted pyrrole JG-03-14, a novel microtubule poison. Biochem. Pharmacol. 2007, 74, 981–991.
- Karatoprak, G.Ş.; Küpeli Akkol, E.; Genç, Y.; Bardakcı, H.; Yücel, Ç.; Sobarzo-Sánchez, E. Combretastatins: An overview of structure, probable mechanisms of action and potential applications. Molecules 2020, 25, 2560.
- Dowlati, A.; Robertson, K.; Cooney, M.; Petros, W.P.; Stratford, M.; Jesberger, J.; Rafie, N.; Overmoyer, B.; Makkar, V.; Stambler, B.; et al. A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin a-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res. 2002, 62, 3408–3416.
- Simoni, D.; Romagnoli, R.; Baruchello, R.; Rondanin, R.; Rizzi, M.; Pavani, M.G.; Alloatti, D.; Giannini, G.; Marcellini, M.; Riccioni, T.; et al. Novel combretastatin analogues endowed with antitumor activity. J. Med. Chem. 2006, 49, 3143–3152.
- West, C.M.; Price, P. Combretastatin A4 phosphate. Anticancer. Drugs 2004, 15, 179–187.
- Rustin, G.J.; Galbraith, S.M.; Anderson, H.; Stratford, M.; Folkes, L.K.; Sena, L.; Gumbrell, L.; Price, P.M. Phase I clinical trial of weekly combretastatin A4 phosphate: Clinical and pharmacokinetic results. J. Clin. Oncol. 2003, 21, 2815–2822.
- Hoang, V.C.; Chow, A.; Emmenegger, U. Abstract 1688: Autophagy inhibition enhances the antitumor effects of combretastatin A4 phosphate (CA4P). Cancer Res. 2013, 73 (Suppl. S8), 1688.
- Greene, L.M.; Meegan, M.J.; Zisterer, D. Combretastatins: More Than Just Vascular Targeting Agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227.
- Agrawal, K. Vinblastine. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4.
- Lee, C.-T.; Huang, Y.-W.; Yang, C.-H.; Huang, K.-S. Drug delivery systems and combination therapy by using vinca alkaloids. Curr. Top. Med. Chem. 2015, 15, 1491–1500.
- Downing, K.H. Structural Basis for the Interaction of Tubulin with Proteins and Drugs that Affect Microtubule Dynamics. Annu. Rev. Cell Dev. Biol. 2000, 16, 89–111.
- Silverman, J.A.; Deitcher, S.R. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother. Pharmacol. 2013, 71, 555–564.
- Moudi, M.; Go, R.; Yien, C.Y.S.; Nazre, M. Vinca alkaloids. Int. J. Prev. Med. 2013, 4, 1231–1235.
- Silvestri, R. New prospects for vinblastine analogues as anticancer agents. J. Med. Chem. 2013, 56, 625–627.
- Krause, W. Resistance to anti-tubulin agents: From vinca alkaloids to epothilones. Cancer Drug Resist. 2019, 2, 2–106.
- Carlson, R.O. New tubulin targeting agents currently in clinical development. Expert Opin. Investig. Drugs 2008, 17, 707–722.
- Schiff, P.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667.
- Horwitz, S.B. Mechanism of action of taxol. Trends Pharmacol. Sci. 1992, 13, 134–136.
- A Jordan, M.; Toso, R.J.; Thrower, D.; Wilson, L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. 1993, 90, 9552–9556.
- Raveendran, R.S.; Baby, S. Resistance to intervention: Paclitaxel in breast cancer. Mini-Rev. Med. Chem. 2021, 21, 1237–1268.
- Baird, R.D.; Tan, D.S.P.; Kaye, S.B. Weekly paclitaxel in the treatment of recurrent ovarian cancer. Nat. Rev. Clin. Oncol. 2010, 7, 575–582.
- Chen, Q.; Xu, S.; Liu, S.; Wang, Y.; Liu, G. Emerging nanomedicines of paclitaxel for cancer treatment. J. Control. Release 2022, 342, 280–294.
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. J. Am. Med. Assoc. 2021, 326, 851–862.
- Zou, C.-F.; Jia, L.; Jin, H.; Yao, M.; Zhao, N.; Huan, J.; Lu, Z.; Bast, R.C.; Feng, Y.; Yu, Y. Re-expression of ARHI (DIRAS3) induces autophagy in breast cancer cells and enhances the inhibitory effect of paclitaxel. BMC Cancer 2011, 11, 22.
- Veldhoen, R.A.; Banman, S.L.; Hemmerling, D.R.; Odsen, R.; Simmen, T.; Simmonds, A.J.; Underhill, D.A.; Goping, I.S. The chemotherapeutic agent paclitaxel inhibits autophagy through two distinct mechanisms that regulate apoptosis. Oncogene 2013, 32, 736–746.
- Gerth, K.; Bedorf, N.; Höfle, G.; Irschik, H.; Reichenbach, H. Epothilons A and B: Antifungal and cytotoxic compounds from Sorangium cellulosum (Myxobacteria). Production, physico-chemical and biological properties. J. Antibiot. 1996, 49, 560–563.
- Nettles, J.H.; Downing, K.H. The binding mode of epothilone A on alpha, beta-tubulin by electron crystallography. Science 2004, 305, 866–869.
- Chou, T.-C.; Zhang, X.-G.; Harris, C.R.; Kuduk, S.D.; Balog, A.; Savin, K.A.; Bertino, J.R.; Danishefsky, S.J. Desoxyepothilone B is curative against human tumor xenografts that are refractory to paclitaxel. Proc. Natl. Acad. Sci. USA 1998, 95, 15798–15802.
- Fumoleau, P.; Coudert, B.; Isambert, N.; Ferrant, E. Novel tubulin-targeting agents: Anticancer activity and pharmacologic profile of epothilones and related analogues. Ann. Oncol. 2007, 18 (Suppl. S5), v9–v15.
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res. 1999, 59, 653–660.
- West, L.M.; Northcote, P.T.; Battershill, C.N. Peloruside A: A Potent Cytotoxic Macrolide Isolated from the New Zealand Marine Sponge Mycale sp. J. Org. Chem. 2000, 65, 445–449.
- Gaitanos, T.N.; Buey, R.M.; Díaz, J.F.; Northcote, P.T.; Teesdale-Spittle, P.; Andreu, J.M.; Miller, J.H. Peloruside A does not bind to the taxoid site on beta-tubulin and retains its activity in multidrug-resistant cell lines. Cancer Res. 2004, 64, 5063–5067.
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.H.; Miller, J.H.; Díaz, J.F.; Steinmetz, M.O. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew. Chem. Int. Ed. Engl. 2014, 53, 1621–1625.
- Castro-Alvarez, A.; Pineda, O.; Vilarrasa, J. Further insight into the interactions of the cytotoxic macrolides laulimalide and peloruside a with their common binding site. ACS Omega 2018, 3, 1770–1782.
- Gollner, A.; Altmann, K.-H.; Gertsch, J.; Mulzer, J. The laulimalide family: Total synthesis and biological evaluation of neolaulimalide, isolaulimalide, laulimalide and a nonnatural analogue. Chem. A Eur. J. 2009, 15, 5979–5997.
- Pickard, R.D.; Spencer, B.H.; McFarland, A.J.; Bernaitis, N.; Davey, A.K.; Perkins, A.V.; Chess-Williams, R.; McDermott, C.M.; Forbes, A.; Christie, D.; et al. Paradoxical effects of the autophagy inhibitor 3-methyladenine on docetaxel-induced toxicity in PC-3 and LNCaP prostate cancer cells. Naunyn-Schmiedebergs Arch. Fur Exp. Pathol. Und Pharmakol. 2015, 388, 793–799.