Epithelial ovarian cancer (EOC) is the deadliest gynecological cancer, and the major cause of death is attributed to metastasis. EOC metastasizes mainly through the transcoelomic pathway, in which cells disseminate from the primary EOC tumor by undergoing epithelial-to-mesenchymal transition (EMT), float freely as spheroids in the ascitic fluid, and then attach onto the mesothelium lining or invade deeper into the peritoneal organs. In addition, metastatic EOC cells can transit in the blood or lymph vessels and undergo extravasation to establish new tumors in hematogenous and lymphatic metastasis. MicroRNAs (miRNAs) are a group of small non-coding RNAs that exert important regulatory functions in many biological processes through their effects on regulating gene expression. Numerous studies have reported that miRNAs can suppress or promote EOC metastasis by regulating the expression of genes involved in various events related to metastasis, such as EMT, cell migration and invasion, as well as tumor angiogenesis and immune suppression.
- epithelial ovarian cancer
- metastasis
- microRNAs (miRNAs)
1. Introduction
Ovarian cancer is the fifth leading cause of cancer-related deaths in females [1]. Based on the cell origin where ovarian tumors arise, ovarian cancer is classified into three categories: epithelial, germ cell, and stromal ovarian cancer. Several types of extremely rare ovarian cancer, such as small cell carcinoma and sarcomas, have also been reported [2]. Among them, epithelial ovarian cancer (EOC) accounts for more than 85% of ovarian cancer cases and is responsible for most ovarian cancer-related deaths [3]. EOC is further grouped into five different histological subtypes, including high-grade serous carcinomas (HGSC), low-grade serous carcinomas (LGSC), endometrioid carcinomas (EC), clear cell carcinomas (CCC), and mucinous carcinomas (MC) [3]. Though the morbidity of ovarian cancer is lower than that in endometrial and cervical cancers, it has the highest mortality rate among gynecological cancers [1]. The five-year survival rate of EOC is less than 45% [4], and relapse and poor prognosis occur in 80% of patients with advanced stages [5][6]. EOC is difficult to detect at the early stages as there are no effective screening methods and the presenting symptoms are vague. Therefore, patients are often diagnosed at the advanced stages when the tumor metastasis is already taking place [5].
MicroRNAs (miRNAs) are small non-coding RNAs that regulate gene expression within cells [7][8]. Studies have shown that 30–60% of human protein-coding genes are regulated by miRNAs [9]. Through regulation of the target gene expression, miRNAs are reported to control many biological processes, including proliferation, differentiation, cell cycle progression, apoptosis, and immune response [10]. Aberrant expression of miRNAs is implicated in many diseases, including cancer. Studies have demonstrated that miRNAs are involved in the progression of EOC [11][12]. Their levels are up- or down-regulated in EOC tumors and/or patient plasma samples, and their abnormal expression is highly associated with EOC metastasis [11][13]. In this review, we provide a brief overview of the biogenesis and mechanisms of actions of miRNAs and metastasis in EOC. We then discuss the dysregulation of miRNAs in EOC and the roles of miRNAs in promoting or suppressing cellular processes related to metastasis. Finally, we point out some limitations of current studies and suggest future research directions.
2. Ovarian Cancer Metastasis
Metastasis is a complex multistep process in which cancer cells disseminate from primary tumors and start new tumors at different sites in the body. This process is regulated by a specific set of genes and signaling pathways. EOC cells mainly metastasize through the transcoelomic pathway [14], in which cells disseminate from the primary EOC tumor by undergoing epithelial-to-mesenchymal transition (EMT) [14] and float freely as spheroids in the ascitic fluid within the peritoneal cavity. The metastatic cells then attach onto the mesothelium lining or invade deeper into the peritoneal organs [15]. In addition, metastatic ovarian cancer cells can transit in the blood or lymph vessels and undergo extravasation to establish new tumors in hematogenous and lymphatic metastasis [15][16]. EOC metastasis to secondary sites accounts for approximately 90% of all ovarian cancer deaths [15]. Therefore, understanding the underlying mechanisms of EOC metastasis could lead to the development of more effective therapeutic tools.
EMT is a biological process which is activated during normal embryonic and organ development, as well as tissue repair [17]. The role of EMT in tumor metastasis has been established in many types of cancers, including EOC [18][19][20]. In EMT, epithelial cells undergo phenotypic alterations through the loss of cell polarity, cell–cell attachment, and gain mesenchymal phenotypes, such as fibroblastoid morphology with increased migratory and invasive properties. EMT is a critical step in ovarian cancer metastasis [21]. Downregulation of epithelial cadherin (E-cadherin, CDH1) and upregulation of mesenchymal neural cadherin (N-cadherin, CDH2) are key elements of EMT. E-cadherin is a transmembrane glycoprotein that associates with β-catenin at the adherens junctions [21]. Loss of E-cadherin results in the destabilization of adherens junctions, promoting cell migration, invasion, and metastasis. E-cadherin expression is repressed directly by many transcription factors, including Snail (SNAI1), Slug (SNAI2), and zinc finger E-box binding homeobox (ZEB)1 and ZEB2, and indirectly by TWIST and TCF4 [22][23]. In addition, Vimentin (VIM), a component of intermediate filaments, is abundantly expressed in mesenchymal cells [24] and exerts inhibitory effects on E-cadherin expression, and cell–cell adhesion, while promoting cell migration and invasion [25]. Therefore, Vimentin is not only an EMT marker but also directly promotes EMT in EOC.
In EOC, EMT is induced by several signaling pathways, including transforming growth factor- β (TGF-β)/Smads, Wnt/β-catenin, PI3K/AKT, Hedgehog, Sonic, and Notch [26]. Wnt signaling promotes the localization of β-catenin into the nucleus, which, in turn, interacts with T-cell factors (TCF/LEF) to regulate transcription [27]. The pathway inhibits E-cadherin by promoting the expression of E-cadherin repressors, such as Snail, Slug, and TWIST [27][28]. TGF-β also enhances EMT through its downstream mediators, SMAD2, SMAD3, and SMAD4 [29]. In addition, the MAPK and PI3K/AKT pathways, activated by many growth factors, or through cross-talks with other signaling molecules, also play critical roles in promoting EMT. For example, epidermal growth factor (EGF) signals through the ERK1/2 and PI3K/AKT pathways to induce EMT [30]. Hepatocyte growth factor (HGF) acts through its receptor, c-Met, and enhances EMT by activating multiple signaling pathways, including MAPK, Wnt/β-catenin, and PI3K/AKT [31][32][33]. Hedgehog glioma-associated oncogene1 (Shh-Gli1) positively regulates EMT via crosstalk with PI3K-AKT [34]. In addition to functions in mitotic progression, Aurora kinase A (AURKA) has been reported to regulate EOC cell migration and invasion in vitro and in vivo [35]. Treatments with AUKA inhibitors, such as alisertib, inhibited migration, adhesion, and EMT via the PI3K/AKT/mTOR- and Sirtuin-1-mediated pathways [35][36], suggesting a potential therapeutic advancement in controlling EOC dissemination. Finally, focal adhesion kinase (FAK) is an important component of various pro-metastatic signaling pathways which promote cancer metastasis, including cell motility [37], cell survival [38][39], invasion [40][41], and EMT [42]. Increased FAK levels are found in several cancers, including EOC [41][42]. In addition, FAK activation, which is determined by p-FAK, increases with tumor progression [42].
Actin filament dynamics are regulated strictly to maintain cell shape and control cell motility [15]. The increase in EOC cell mobility is mediated by actin filament remodeling via the activation of GTPase signaling pathways. For example, GTPase RAP1B has been reported to activate Src and JNK to facilitate integrin-mediated actin remodeling and thereby promote metastasis [43]. DAAM1, which is upregulated in EOC tumors, activates RHOA, induces the formation of microfilaments, and promotes cell migration and invasion [44]. In addition, Lim kinase 1 (LIMK1), a member of serine-threonine protein kinases that acts downstream of RHO GTPase signaling, also participates in actin remodeling in EOC [45]. LIMK1 is a key player in the reorganization of the actin cytoskeleton by inactivating actin-binding factor cofilin through phosphorylation [46]. LIMK1 protein levels are upregulated in EOC and correlated with poor differentiation [45]. In addition, knockout of LIMK1 inhibited migration and invasion of EOC cells [45], supporting its role in promoting EOC cell mobility.
Most EOC metastasis occurs in the peritoneal cavity. Once escaping the primary site, ovarian tumor cells transit in the ascitic fluid as single cells or aggregated cells, referred to as spheroids, and exhibit cancer stem-like properties [47][48]. Cancer cells then adhere to the mesothelium lining of the peritoneum through the binding of integrin receptors to the extracellular matrix (ECM) elements of the mesothelial cells [15]. The integrin-ECM interaction was suggested to activate integrin-linked kinase (ILK) through phosphorylation, promoting a phosphorylation cascade of a variety of ILK-intracellular substrates, including protein kinase B (PKB/AKT), glycogen synthase kinase-3 (GSK-3), and myosin light chain at focal adhesions, and promoting cell adhesion and invasion to the mesothelium [49]. In addition, ovarian tumor cells increase the production of proteolytic enzymes, such as matrix metalloproteases (MMPs), which recognize and degrade ECM elements, enhancing invasive behavior. MMPs play a role in EMT and they are also activated by genes and signaling pathways that induce EMT [50]. In EOC, it has been reported that knockdown of SNAI1 reduced MMP2 but upregulated its inhibitor, TIMP2, suggesting that Snail induces MMP activity [51]. Moreover, EOC cells avoid apoptosis while detaching from primary sites and circulating in ascites or transiting to a distant location by resisting anoikis, a programmed cell death which is activated to inhibit anchorage-independent growth or cell adhesion to an inappropriate matrix [52]. Among steps that occur in cancer metastasis, escaping apoptosis is critical in tumor development and metastasis [53].
Interaction between cancer cells and the tumor environment also plays a role in metastasis. Hypoxia is commonly observed in fast-growing tumors with an insufficient supply of oxygen. Under hypoxic conditions, the association of stabilized hypoxia-inducible factor (HIF)-1α and HIF-2α [54] with HIF-1β induces the expression of downstream target genes that are involved in cell invasion, and metastasis [55]. LOX, one of the target genes induced by HIF-1 complex, has been shown to cross-link collagen and provide a linear track for cell migration [56][57]. In addition, HIF-1 complex modulates the downregulation of DMN2, resulting in decreased endocytosis, an energy-consuming cellular process [56]. Hypoxia has also been reported to down-regulate BRCA1 expression via Retinoblastoma-associated protein E2F transcription factor and suppresses homologous recombination in hypoxic cancer cells, potentially increasing genomic instability [58][59]. Furthermore, the behaviors of metastatic EOC cells are influenced by secreted factors residing in ascites. Cytokine CXCL12 and hyaluronic acid in ascitic fluid have been demonstrated to interact with CXCR4 and CD44 receptors on EOC cell surface respectively, stimulating cell migration, angiogenesis, and localization to the peritoneal surface [15][60][61][62].
Lastly, the metastasis of EOC cells is enhanced by an immunosuppressive microenvironment. Tumor-infiltrating lymphocytes (TILs), such as T cells, B cells, macrophages, and natural killer cells, were also found to be present in ascites and pelvic peritoneal biopsies of advanced ovarian cancer patients [63]. Among them, tumor-associated macrophages (TAMs) play a role in the suppression of adaptive immunity. TAMs induced the imbalance of Treg/Th17 and promoted angiogenesis and metastasis via cross-talk with endothelial cells in EOC [64][65]. In addition, TIL-produced cytokines, such as IL-6, IL-10, ARG-1, and CCL-2, have been reported to promote tumor progression and metastasis, and are involved in immune subversion [66][67]. In addition, EOC cells promote immune evasion via downregulating tumor-associated surface ligands. MHC class I chain-related molecules A and B (MICA and MICB) are widely expressed on epithelial tumor cells and targeted by cytotoxic lymphocytes such as CD8+ T cells and natural killer (NK) cells [68]. Downregulation and internalization of MICA/B have been reported in EOC [68][69], allowing EOC malignant cells to escape immune surveillance.
3. Roles of miRNAs in Ovarian Cancer Metastasis
Numerous studies have reported the functions of miRNAs in EMT, cell migration, invasion, and metastasis in EOC. In addition, miRNAs also participate in inducing angiogenesis and modulating tumor microenvironments [64][67][68], which contribute to tumor metastasis (Figure 1). The majority of studied miRNAs exert negative regulatory effects on metastasis, while some miRNAs serve as positive regulators of metastasis. In addition, some miRNAs have been reported to exhibit both pro-metastatic and anti-metastatic effects, probably depending on the genes they targeted under different cancer contexts.
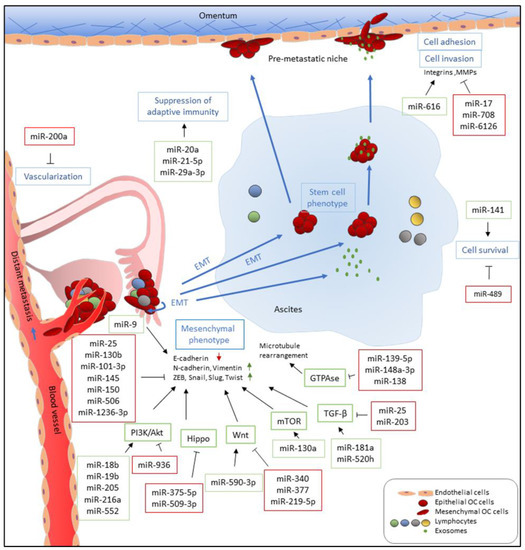
Figure 1. Functions of miRNAs in EOC metastasis. miRNAs which are upregulated and promote metastasis-related processes are depicted in green boxes while downregulated miRNAs which inhibit metastatic-related processes are listed in red boxes. miRNAs directly and indirectly regulate epithelial-to-mesenchymal transition (EMT) by targeting E-cadherin and E-cadherin repressors, such as ZEB, Snail, Slug, and Twist, and associated signaling pathways. miR-9 directly targets E-cadherin to activate EMT while miR-25, miR-101-3p, miR-130b, miR-145, miR-150, and miR-1236-3b directly inhibit expression of E-cadherin repressors, including ZEB1, SNAI2, and TWIST1. Multiple miRNAs, such as miR-18b, miR-19b, miR-205, miR-216b, and miR-552, promote EMT by targeting PTEN, leading to the activation of the PI3K/AKT pathway. In addition, upregulation of miR-590-3p, miR-130a, miR-181a, and miR-520h induces Wnt, mTOR, and TGF-β signaling, respectively, which are known pathways that promote EMT. In contrast, miR-936, miR-375-5p, miR-509-3p, miR-340, miR-377, miR-219-5p, miR-25, and miR-203 attenuate PI3K/AKT, Hippo, Wnt, and TGF-β signaling pathways and inhibit cell migration, invasion, and EMT. Furthermore, downregulation of miR-139-5p, miR-148a-3p, and miR-138, which has been reported to inhibit ROCKs and LIMK1 expression, increases cell motility via GTPase signaling. Metastatic EOC cells float in ascites as single cells or spheroids which exhibit stem cell-like properties. To survive after detaching from primary site and inside ascites, metastatic EOC cells upregulate miR-141 and downregulate miR-489 to modulate anoikis resistance. In the transcoelomic pathway, EOC metastatic cells then adhere to the mesothelium lining and invade peritoneal organs. Upregulation of miR-616 and downregulation of miR-17 and miR-6126 increase cell adhesion via increased expression and activities of integrins and MMPs which recognize and degrade the extracellular matrix (ECM) of the mesothelial cells, respectively. In addition, downregulation of miR-708 increases focal adhesion formation through promoting focal adhesion kinase (FAK) activities. EOC metastasis occurs in an immune-suppressive environment which are modulated by miRNAs. miR-21-5p and miR-29a-3p promote adaptive immune suppression via upregulation of tumor-associated macrophages (TAMs) and induction Treg/Th17 imbalance while miR-20a downregulates MICA/B to avoid recognition by cytotoxic T-cells. Furthermore, EOC tumors increase vascularization and angiogenesis via downregulation of miR-200a, implicating them in distant metastasis through a perfusion pathway.
4. Conclusions and Future Direction
Cancer metastasis is one of the main factors that leads to poor clinical outcomes for EOC patients. Accumulating evidence demonstrates that miRNAs play important roles in EOC metastasis. Aberrant expression of miRNAs has been reported in EOC. Such dysregulation can be attributed to alterations at the DNA level, such as amplification and hypermethylation at the promoter regions of miRNA genes. In addition, altered transcriptional controls and defects in miRNA biogenesis machinery also contribute to the abnormality of miRNA levels. Many studies have reported that miRNA expression profiles correlate with clinical features, such as tumor stage, grade, and overall survival of patients, raising the possibility of using miRNAs as diagnostic and/or prognostic markers.
Due to the heterogeneity of EOC, it is a challenge to find effective biomarkers for detecting EOC in different tumors [70]. miRNA profiling studies have sometimes reported inconsistent findings. One of the underlying issues could be the controls used. Some researchers used normal ovarian tissues as the control while others used benign tumors. Methylation patterns of some miRNA genes have been shown to be correlated with metastasis [71][72]. In addition, miRNAs are detected in biological fluids, which can serve as a non-invasive tool for EOC diagnosis. Further efforts in validating the specificity and sensitivity of miRNA signatures in a large cohort of EOC patients are needed for the development of miRNAs as diagnostic and prognostic biomarkers.
Metastasis of ovarian cancer is orchestrated by several interconnected biological processes, including EMT, increase in cell migration and invasion, destruction of the ECM, formation of spheroids, avoidance of apoptosis, angiogenesis, and immune suppression [53]. Some miRNAs have been reported to promote metastasis, mainly by targeting negative regulators of these processes. On the other hand, most miRNAs that have been studied exert suppressive effects on metastasis, mainly by inhibiting transcription factors that induce the expression of mesenchymal markers, or key signaling pathways that promote EMT, motility, and tumor angiogenesis. Therefore, the upregulation of metastasis-promoting miRNAs and downregulation of metastasis-suppressing miRNAs would lead to a dysregulated signaling network and promote metastasis. However, more work is required to better understand the role of miRNAs and the underlying mechanisms by which they regulate metastasis. Among the studies reported so far, some are comprehensive, but most only examined the effects of miRNAs using established EOC cell lines in vitro. Therefore, further in vivo experiments would verify the roles of those miRNAs in metastasis of EOC. Moreover, miRNAs can target many genes and it is possible that they could exert tumor-promoting or tumor-suppressive effects, depending on the relative abundance and/or functions of target genes in different tumor contexts. Most studies have been focused on one or a few target genes. Additional studies in identifying critical targets that are directly involved in the induction of EOC metastasis would enhance our understanding of the roles of miRNAs in these processes. Finally, EOC consists of multiple histological subtypes, each one with unique origins and distinct molecular features [3]. More work on examining the dysregulation and functions of miRNAs in different subtypes of EOC is warranted as it may help to develop precise therapeutics. miRNAs have been suggested as promising therapeutic targets for cancer treatment [73]. miRNA-based therapies have been established for lung cancer treatment and further trials are anticipated to address clinical treatment efficacy [73]. It is possible that the restoration of down-regulated miRNAs that inhibit metastasis or inhibition of up-regulated miRNAs that promote metastasis could be used in the future as a therapeutic approach for EOC.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21197093
References
- Siegel, R. L.; Miller, K. D.; Jemal, A., Cancer statistics, 2019. CA Cancer J Clin 2019, 69 (1), 7-34.
- Boussios, S.; Moschetta, M.; Zarkavelis, G.; Papadaki, A.; Kefas, A.; Tatsi, K., Ovarian sex-cord stromal tumours and small cell tumours: Pathological, genetic and management aspects. Crit Rev Oncol Hematol 2017, 120, 43-51.
- National Academies of Sciences, E., and Medicine, Ovarian Cancers: Evolving Paradigms in Research and Care. The National Academies Press: Washington, DC, 2016; p 396.
- Lupia, M.; Cavallaro, U., Ovarian cancer stem cells: still an elusive entity? Molecular cancer 2017, 16 (1), 64.
- Webb, P. M.; Jordan, S. J., Epidemiology of epithelial ovarian cancer. Best Pract Res Clin Obstet Gynaecol 2017, 41, 3-14.
- Matulonis, U. A.; Sood, A. K.; Fallowfield, L.; Howitt, B. E.; Sehouli, J.; Karlan, B. Y., Ovarian cancer. Nat Rev Dis Primers 2016, 2, 16061.
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T., Identification of novel genes coding for small expressed RNAs. Science 2001, 294 (5543), 853-8.
- Ambros, V., microRNAs: tiny regulators with great potential. Cell 2001, 107 (7), 823-6.
- Friedman, R. C.; Farh, K. K.; Burge, C. B.; Bartel, D. P., Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009, 19 (1), 92-105.
- Vidigal, J. A.; Ventura, A., The biological functions of miRNAs: lessons from in vivo studies. Trends Cell Biol 2015, 25 (3), 137-47.
- Staicu, C. E.; Predescu, D. V.; Rusu, C. M.; Radu, B. M.; Cretoiu, D.; Suciu, N.; Cretoiu, S. M.; Voinea, S. C., Role of microRNAs as Clinical Cancer Biomarkers for Ovarian Cancer: A Short Overview. Cells 2020, 9 (1).
- Chen, S. N.; Chang, R.; Lin, L. T.; Chern, C. U.; Tsai, H. W.; Wen, Z. H.; Li, Y. H.; Li, C. J.; Tsui, K. H., MicroRNA in Ovarian Cancer: Biology, Pathogenesis, and Therapeutic Opportunities. Int J Environ Res Public Health 2019, 16 (9).
- Roberts, C. M.; Cardenas, C.; Tedja, R., The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers 2019, 11 (8).
- Tan, D. S.; Agarwal, R.; Kaye, S. B., Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol 2006, 7 (11), 925-34.
- Yeung, T. L.; Leung, C. S.; Yip, K. P.; Au Yeung, C. L.; Wong, S. T.; Mok, S. C., Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am J Physiol Cell Physiol 2015, 309 (7), C444-56.
- Lengyel, E., Ovarian cancer development and metastasis. The American journal of pathology 2010, 177 (3), 1053-64.
- Micalizzi, D. S.; Farabaugh, S. M.; Ford, H. L., Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia 2010, 15 (2), 117-34.
- Onder, T. T.; Gupta, P. B.; Mani, S. A.; Yang, J.; Lander, E. S.; Weinberg, R. A., Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 2008, 68 (10), 3645-54.
- Mani, S. A.; Yang, J.; Brooks, M.; Schwaninger, G.; Zhou, A.; Miura, N.; Kutok, J. L.; Hartwell, K.; Richardson, A. L.; Weinberg, R. A., Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A 2007, 104 (24), 10069-74.
- Davidson, B.; Trope, C. G.; Reich, R., Epithelial-mesenchymal transition in ovarian carcinoma. Front Oncol 2012, 2, 33.
- Palma Flores, C.; Garcia-Vazquez, R.; Gallardo Rincon, D.; Ruiz-Garcia, E.; Astudillo de la Vega, H.; Marchat, L. A.; Salinas Vera, Y. M.; Lopez-Camarillo, C., MicroRNAs driving invasion and metastasis in ovarian cancer: Opportunities for translational medicine (Review). Int J Oncol 2017, 50 (5), 1461-1476.
- Peinado, H.; Olmeda, D.; Cano, A., Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature reviews. Cancer 2007, 7 (6), 415-28.
- Yang, J.; Weinberg, R. A., Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Developmental cell 2008, 14 (6), 818-29.
- Tsai, J. H.; Yang, J., Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes & development 2013, 27 (20), 2192-206.
- Sun, Y.; Hu, L.; Zheng, H.; Bagnoli, M.; Guo, Y.; Rupaimoole, R.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Ji, P.; Chen, K.; Sood, A. K.; Mezzanzanica, D.; Liu, J.; Sun, B.; Zhang, W., MiR-506 inhibits multiple targets in the epithelial-to-mesenchymal transition network and is associated with good prognosis in epithelial ovarian cancer. J Pathol 2015, 235 (1), 25-36.
- Kalluri, R.; Weinberg, R. A., The basics of epithelial-mesenchymal transition. The Journal of clinical investigation 2009, 119 (6), 1420-8.
- Nusse, R.; Clevers, H., Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169 (6), 985-999.
- Nguyen, V. H. L.; Hough, R.; Bernaudo, S.; Peng, C., Wnt/beta-catenin signalling in ovarian cancer: Insights into its hyperactivation and function in tumorigenesis. J Ovarian Res 2019, 12 (1), 122.
- Rafehi, S.; Ramos Valdes, Y.; Bertrand, M.; McGee, J.; Prefontaine, M.; Sugimoto, A.; DiMattia, G. E.; Shepherd, T. G., TGFbeta signaling regulates epithelial-mesenchymal plasticity in ovarian cancer ascites-derived spheroids. Endocr Relat Cancer 2016, 23 (3), 147-59.
- Zhou, X.; Hu, Y.; Dai, L.; Wang, Y.; Zhou, J.; Wang, W.; Di, W.; Qiu, L., MicroRNA-7 inhibits tumor metastasis and reverses epithelial-mesenchymal transition through AKT/ERK1/2 inactivation by targeting EGFR in epithelial ovarian cancer. PLoS One 2014, 9 (5), e96718.
- Moran-Jones, K., The Therapeutic Potential of Targeting the HGF/cMET Axis in Ovarian Cancer. Mol Diagn Ther 2016, 20 (3), 199-212.
- Zhang, R.; Shi, H.; Ren, F.; Feng, W.; Cao, Y.; Li, G.; Liu, Z.; Ji, P.; Zhang, M., MicroRNA-338-3p suppresses ovarian cancer cells growth and metastasis: implication of Wnt/catenin beta and MEK/ERK signaling pathways. Journal of experimental & clinical cancer research : CR 2019, 38 (1), 494.
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; Zeng, Z.; Xiong, W., Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Molecular cancer 2018, 17 (1), 45.
- Ke, Z.; Caiping, S.; Qing, Z.; Xiaojing, W., Sonic hedgehog-Gli1 signals promote epithelial-mesenchymal transition in ovarian cancer by mediating PI3K/AKT pathway. Medical oncology (Northwood, London, England) 2015, 32 (1), 368.
- Do, T. V.; Xiao, F.; Bickel, L. E.; Klein-Szanto, A. J.; Pathak, H. B.; Hua, X.; Howe, C.; O'Brien, S. W.; Maglaty, M.; Ecsedy, J. A.; Litwin, S.; Golemis, E. A.; Schilder, R. J.; Godwin, A. K.; Connolly, D. C., Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene 2014, 33 (5), 539-49.
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N., Wise Management of Ovarian Cancer: On the Cutting Edge. J Pers Med 2020, 10 (2).
- Mitra, S. K.; Hanson, D. A.; Schlaepfer, D. D., Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 2005, 6 (1), 56-68.
- Cance, W. G.; Golubovskaya, V. M., Focal adhesion kinase versus p53: apoptosis or survival? Sci Signal 2008, 1 (20), pe22.
- Frisch, S. M.; Schaller, M.; Cieply, B., Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci 2013, 126 (Pt 1), 21-9.
- Shibue, T.; Brooks, M. W.; Inan, M. F.; Reinhardt, F.; Weinberg, R. A., The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov 2012, 2 (8), 706-21.
- Sood, A. K.; Coffin, J. E.; Schneider, G. B.; Fletcher, M. S.; DeYoung, B. R.; Gruman, L. M.; Gershenson, D. M.; Schaller, M. D.; Hendrix, M. J., Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. The American journal of pathology 2004, 165 (4), 1087-95.
- Zhao, J.; Guan, J. L., Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev 2009, 28 (1-2), 35-49.
- Lin, K. T.; Yeh, Y. M.; Chuang, C. M.; Yang, S. Y.; Chang, J. W.; Sun, S. P.; Wang, Y. S.; Chao, K. C.; Wang, L. H., Glucocorticoids mediate induction of microRNA-708 to suppress ovarian cancer metastasis through targeting Rap1B. Nat Commun 2015, 6, 5917.
- Mei, J.; Huang, Y.; Hao, L.; Liu, Y.; Yan, T.; Qiu, T.; Xu, R.; Xu, B.; Xiao, Z.; Jiang, X.; Hu, K.; Zhu, Y., DAAM1-mediated migration and invasion of ovarian cancer cells are suppressed by miR-208a-5p. Pathol Res Pract 2019, 215 (7), 152452.
- Chen, P.; Zeng, M.; Zhao, Y.; Fang, X., Upregulation of Limk1 caused by microRNA-138 loss aggravates the metastasis of ovarian cancer by activation of Limk1/cofilin signaling. Oncol Rep 2014, 32 (5), 2070-6.
- Prunier, C.; Prudent, R.; Kapur, R.; Sadoul, K.; Lafanechere, L., LIM kinases: cofilin and beyond. Oncotarget 2017, 8 (25), 41749-41763.
- Wang, L.; Mezencev, R.; Svajdler, M.; Benigno, B. B.; McDonald, J. F., Ectopic over-expression of miR-429 induces mesenchymal-to-epithelial transition (MET) and increased drug sensitivity in metastasizing ovarian cancer cells. Gynecol Oncol 2014, 134 (1), 96-103.
- Luo, X.; Dong, Z.; Chen, Y.; Yang, L.; Lai, D., Enrichment of ovarian cancer stem-like cells is associated with epithelial to mesenchymal transition through an miRNA-activated AKT pathway. Cell Prolif 2013, 46 (4), 436-46.
- Persad, S.; Dedhar, S., The role of integrin-linked kinase (ILK) in cancer progression. Cancer Metastasis Rev 2003, 22 (4), 375-84.
- Scheau, C.; Badarau, I. A.; Costache, R.; Caruntu, C.; Mihai, G. L.; Didilescu, A. C.; Constantin, C.; Neagu, M., The Role of Matrix Metalloproteinases in the Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma. Anal Cell Pathol (Amst) 2019, 2019, 9423907.
- Jin, H.; Yu, Y.; Zhang, T.; Zhou, X.; Zhou, J.; Jia, L.; Wu, Y.; Zhou, B. P.; Feng, Y., Snail is critical for tumor growth and metastasis of ovarian carcinoma. Int J Cancer 2010, 126 (9), 2102-11.
- Paoli, P.; Giannoni, E.; Chiarugi, P., Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta 2013, 1833 (12), 3481-3498.
- Braga, E. A.; Fridman, M. V.; Kushlinskii, N. E., Molecular Mechanisms of Ovarian Carcinoma Metastasis: Key Genes and Regulatory MicroRNAs. Biochemistry (Mosc) 2017, 82 (5), 529-541.
- Semenza, G. L., Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci 2012, 33 (4), 207-14.
- Keith, B.; Johnson, R. S.; Simon, M. C., HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nature reviews. Cancer 2011, 12 (1), 9-22.
- Joshi, H. P.; Subramanian, I. V.; Schnettler, E. K.; Ghosh, G.; Rupaimoole, R.; Evans, C.; Saluja, M.; Jing, Y.; Cristina, I.; Roy, S.; Zeng, Y.; Shah, V. H.; Sood, A. K.; Ramakrishnan, S., Dynamin 2 along with microRNA-199a reciprocally regulate hypoxia-inducible factors and ovarian cancer metastasis. Proc Natl Acad Sci U S A 2014, 111 (14), 5331-6.
- Schietke, R.; Warnecke, C.; Wacker, I.; Schodel, J.; Mole, D. R.; Campean, V.; Amann, K.; Goppelt-Struebe, M.; Behrens, J.; Eckardt, K. U.; Wiesener, M. S., The lysyl oxidases LOX and LOXL2 are necessary and sufficient to repress E-cadherin in hypoxia: insights into cellular transformation processes mediated by HIF-1. J Biol Chem 2010, 285 (9), 6658-69.
- Bindra, R. S.; Gibson, S. L.; Meng, A.; Westermark, U.; Jasin, M.; Pierce, A. J.; Bristow, R. G.; Classon, M. K.; Glazer, P. M., Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res 2005, 65 (24), 11597-604.
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N., Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics (Basel) 2019, 9 (3).
- Balkwill, F., Cancer and the chemokine network. Nature reviews. Cancer 2004, 4 (7), 540-50.
- Lv, Y.; Lei, Y.; Hu, Y.; Ding, W.; Zhang, C.; Fang, C., miR-448 negatively regulates ovarian cancer cell growth and metastasis by targeting CXCL12. Clin Transl Oncol 2015, 17 (11), 903-9.
- Casey, R. C.; Skubitz, A. P., CD44 and beta1 integrins mediate ovarian carcinoma cell migration toward extracellular matrix proteins. Clin Exp Metastasis 2000, 18 (1), 67-75.
- Santoiemma, P. P.; Powell, D. J., Jr., Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol Ther 2015, 16 (6), 807-20.
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X., Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol Res 2018, 6 (12), 1578-1592.
- Wang, X.; Zhu, Q.; Lin, Y.; Wu, L.; Wu, X.; Wang, K.; He, Q.; Xu, C.; Wan, X.; Wang, X., Crosstalk between TEMs and endothelial cells modulates angiogenesis and metastasis via IGF1-IGF1R signalling in epithelial ovarian cancer. Br J Cancer 2017, 117 (9), 1371-1382.
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A., Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002, 23 (11), 549-55.
- Jiang, B.; Zhu, S. J.; Xiao, S. S.; Xue, M., MiR-217 Inhibits M2-Like Macrophage Polarization by Suppressing Secretion of Interleukin-6 in Ovarian Cancer. Inflammation 2019, 42 (5), 1517-1529.
- Xie, J.; Liu, M.; Li, Y.; Nie, Y.; Mi, Q.; Zhao, S., Ovarian tumor-associated microRNA-20a decreases natural killer cell cytotoxicity by downregulating MICA/B expression. Cell Mol Immunol 2014, 11 (5), 495-502.
- Ghadially, H.; Brown, L.; Lloyd, C.; Lewis, L.; Lewis, A.; Dillon, J.; Sainson, R.; Jovanovic, J.; Tigue, N. J.; Bannister, D.; Bamber, L.; Valge-Archer, V.; Wilkinson, R. W., MHC class I chain-related protein A and B (MICA and MICB) are predominantly expressed intracellularly in tumour and normal tissue. Br J Cancer 2017, 116 (9), 1208-1217.
- Dochez, V.; Caillon, H.; Vaucel, E.; Dimet, J.; Winer, N.; Ducarme, G., Biomarkers and algorithms for diagnosis of ovarian cancer: CA125, HE4, RMI and ROMA, a review. J Ovarian Res 2019, 12 (1), 28.
- Filippova, E. A.; Loginov, V. I.; Burdennyi, A. M.; Braga, E. A.; Pronina, I. V.; Kazubskaya, T. P.; Kushlinskii, D. N.; Utkin, D. O.; Fridman, M. V.; Khodyrev, D. S.; Kushlinskii, N. E., Hypermethylated Genes of MicroRNA in Ovarian Carcinoma: Metastasis Prediction Marker Systems. Bull Exp Biol Med 2019, 167 (1), 79-83.
- Loginov, V. I.; Pronina, I. V.; Burdennyy, A. M.; Filippova, E. A.; Kazubskaya, T. P.; Kushlinsky, D. N.; Utkin, D. O.; Khodyrev, D. S.; Kushlinskii, N. E.; Dmitriev, A. A.; Braga, E. A., Novel miRNA genes deregulated by aberrant methylation in ovarian carcinoma are involved in metastasis. Gene 2018, 662, 28-36.
- Wu, S. G.; Chang, T. H.; Liu, Y. N.; Shih, J. Y., MicroRNA in Lung Cancer Metastasis. Cancers 2019, 11 (2).