Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
G-quadruplexes (G4) are now extensively recognised as a peculiar non-canonical DNA geometry that plays a prime importance role in processes of biological relevance whose number is increasing continuously. The same is true for the less-studied RNA G4 counterpart. G4s are stable structures; their geometrical parameters may be finely tuned not only by the presence of particular sequences of nucleotides but also by the salt content of the medium or by a small molecule that may act as a peculiar topology inducer.
- selectivity
- G4 topology
- porphyrin
- phenanthroline
1. Introduction
A G-quadruplex (G4) is a four-stranded DNA or RNA structure with stacked guanine tetrads (G-tetrads) held together by eight hydrogen bonds (Figure 1a) [1]. At the end of the XXth century, quadruplex sequences were identified at the end of telomeric DNA in eukaryotic chromosomes [2] and gene promoter regions [3]. These first findings constituted the start of enthusiastic studies on G4s, which became the target for cutting-edge research, in particular for the design and development of novel anticancer drugs. The studies on RNA G4s are more recent, but it is now clear that RNA G4s and G4s formed on RNA-DNA hybrids are also involved in regulating chromosome functions that involve DNA, RNA and RNA/DNA complexes [4][5][6][7]. In the last two decades, researchers searched for molecules that could selectively discriminate between G4s and double-stranded DNA/RNA polynucleotides. However, it quickly became clear that sequences that could fold into G4s could have produced species with a very rich structural polymorphism and that different G4s typologies could be associated with different cellular processes [8][9][10]. Selectivity is a key point for efficient therapies, so the problem moved to enable the differentiation of different types of G4s. Note that polymorphisms need to be carefully considered not only from a static point of view but also in the frame of several geometries connected by equilibria [11]. The major, dominating one will depend not only on geometrical/sequence factors but also on the medium (type and concentration of cations) and on the possible presence of some small molecule, specially designed to switch the reaction towards a peculiar geometry. This aspect, on the one hand, complicates much of the picture but, on the other hand, is fascinating and opens the way to new scientific strategies to reach the goal to find optimised drugs or even using G4 assemblies for obtaining responsive nanomaterials with electronic, biomedical and drug-delivery applications [12].
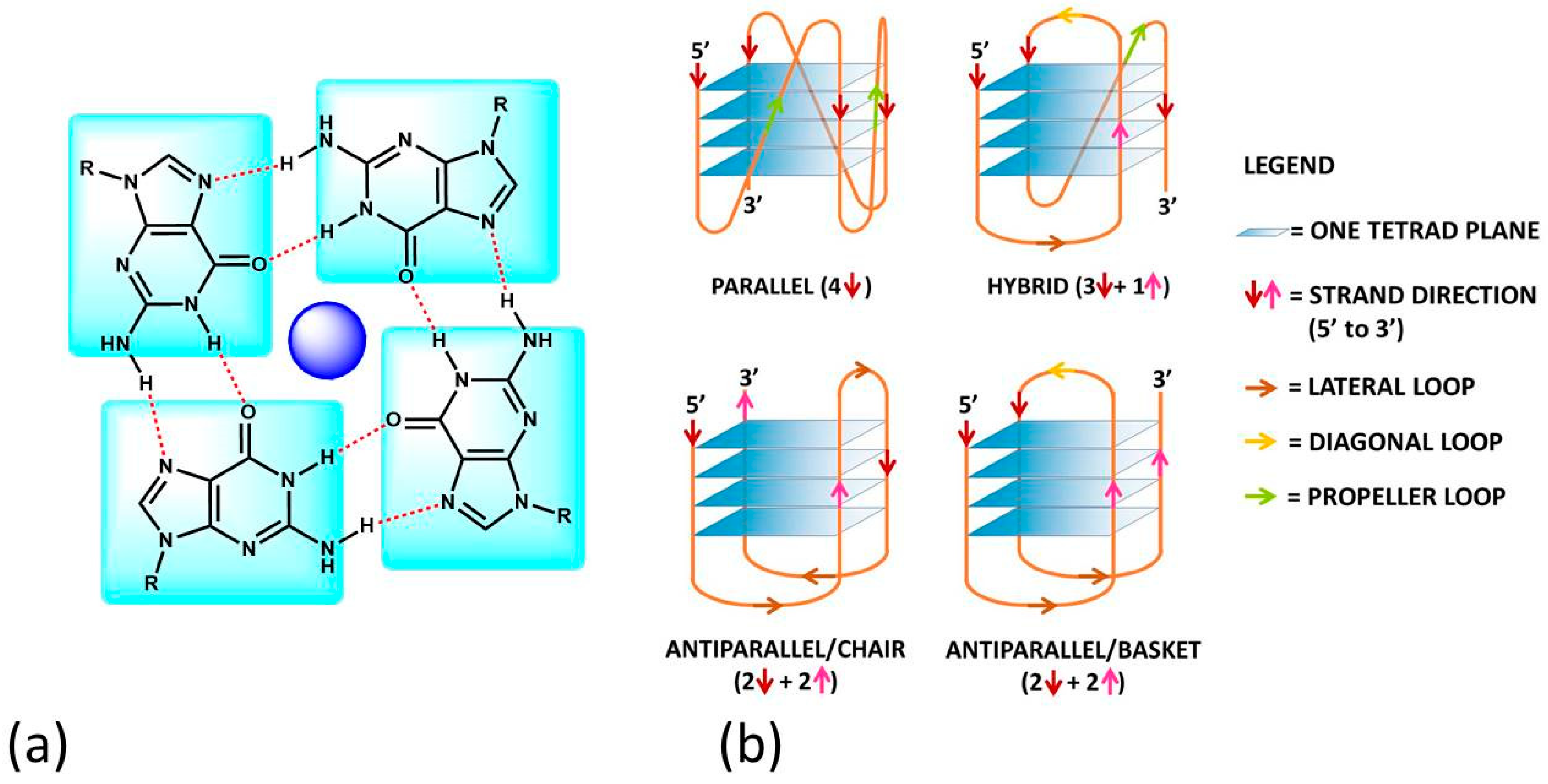
Figure 1. (a) The guanine-tetrad (G4) and (b) examples of structural conformations of G4s. In (b), the intramolecular type is taken into account; the same classifications hold in the intermolecular case. In (b), the oligonucleotide strand goes in the 5′ to 3′ direction; the vertical arrows (red or pink) indicate the direction of the part of the strand involved in G-tetrad formation, and the diagonal arrows refer to the direction of the loops. The latter can be of three types: (i) the lateral loop connects two adjacent guanines of the same G-quartet plane (brown arrow); (ii) the diagonal loop connects two opposite guanines of the same G-quartet plane (yellow arrow) and (iii) the propeller loop connects two guanines belonging to two different (upper and lower) G-quartet planes (green arrow).
G4s can arise from the folding of a single strand (intramolecular G4). The formation of these quadruplex structures in human chromosomes is possible since the terminal nucleotides of telomeric DNA are single-stranded [13]. On the other hand, G4s can be formed by the combination of two or more separated strands (intermolecular G4s). G4s can display a wide variety of topologies (Figure 1b), as a consequence of several possible combinations of strand direction (intended as 5′ → 3′), number of G-tetrads, as well as variations in the size and the disposition of the loops (arising from the nucleotides not involved in the G-tetrads). G4s can be classified as parallel if the strands are all oriented in the same direction (4 ↑ or ↓) [14]. On the contrary, antiparallel structures derive from strands directed in different ways (2↑+ 2↓) [15]; these G4s can be arranged as a chair or basket-type structures depending on the loops’ position. If the structure is instead formed by three parallel and one antiparallel strand, the G4 is defined as a hybrid (3↑ + 1↓ or 3↓ + 1↑) [16].
The formation, stability and conformation of the quadruplexes are dependent on monovalent cations [17]. M+ ions can conveniently fit into the central channel of the G-tetrads, according to the strong negative electrostatic potential created by the guanine oxygen atoms [18]. The precise location of the cation depends on the nature of M+. Na+ ions have been found as centred in the plane of a G-tetrad as well as in the space between two successive G-tetrads; K+ ions are, instead, always equidistant between each G-tetrad plane and form a symmetric tetragonal bipyramidal configuration with the eight oxygen atoms [1]. Because K+ is much more abundant than Na+ in cellular environments, the knowledge of the human telomeric G4 structure in K+ solution is more important. K+ ions have been found to induce hybrid-type conformations for human telomeric G4, whereas Na+ ions tend to promote antiparallel structures [19]. M2+ ions may also play an important role [6]; for instance, Ca2+ will favour parallel structures [20].
As already cited above, ligand–G4 interactions are extensively investigated since G4s are involved in important biological processes, especially concerning cancer cells [21]. Therefore, ligands that stabilize or induce the formation of G4 structures can be considered promising anticancer agents [22][23]. Besides crystals, as for geometries in solution, detailed CD and NMR spectroscopic studies, together with computational investigations [24][25][26][27][28], have provided a clear understanding of quadruplex structures. Additionally, innovative approaches such as 2D IR spectroscopy are proposed for analysing G4 structures [29]. On this basis, it is possible to develop a rational approach to the design of quadruplex binding ligands with potential anticancer activity [30]. Geometrical complementarity plays a major role in the search for high affinity and selectivity. Good G4 binders should present extended π-planar structures able to stack the external G-tetrads. Moreover, positively charged substituents promote the affinity with the grooves and the loops of G4 thanks to the electrostatic attraction with the negatively charged phosphate backbone. Lastly, a partial positive charge, substituting the cationic charge of the potassium or sodium that would normally occupy the G4 centre, can increase adduct stabilisation [31]. Optimal ligands should be selective for G4 over double-helix DNA since the double-stranded helix constitutes the major component of the human genome and its binding can prelude general cellular toxicity [32]. G4 selectivity can arise, at least in part, from the difference between the large, highly accessible surface area of a terminal quartet compared with the much smaller, less accessible G-C or A-T base pair surfaces of a typical duplex DNA [33]. Common G4 binders are macrocyclic ligands such as porphyrins, phthalocyanines and their metal complexes [34]. Meso-5,10,15,20-Tetrakis-(N-methyl-4-pyridyl)porphine (TMPyP4) represents the most commonly studied example for the porphyrin class [35]. Metal–salphen and metal–salen complexes have shown strong affinity and high selectivity for G4s as well [36]. Square-planar Ni(II)–salphen complexes have been found to inhibit telomerase activity by stabilizing G4 structures [37]. Aromatic molecules that bear charged peripheral chains (such as perylene diimide and phenanthroline derivatives) can bind G4 through the π-stacking interaction between the aromatic core and the G-tetrads along with electrostatic interactions between the substituents and the G4 backbone [38]. Investigations on non-planar metal complexes are rarer, but some examples have been reported in the literature anyway. For example, the binding properties of dinuclear [Ru(II)(phen)2(dppz)2+] complexes have been successfully investigated [39]. Concerning the binding position of the ligand, the intercalation of small molecules between the quadruplex tetrads is thought to be difficult because G4s are extremely stable and rigid. Besides, Figure 1a enlightens the presence of eight hydrogen bonds in one G-tetrad, i.e., more than the two or three hydrogen bonds present in each AT or GC base pairing, respectively. Accordingly, any possible G4 distortion requires a very high energy cost [40]. Thus, the stacking of the ligand on the outer surface of G4 as well as its disposition on the grooves appear to be more energetically favourable and probable binding modes (Figure 2) [8].
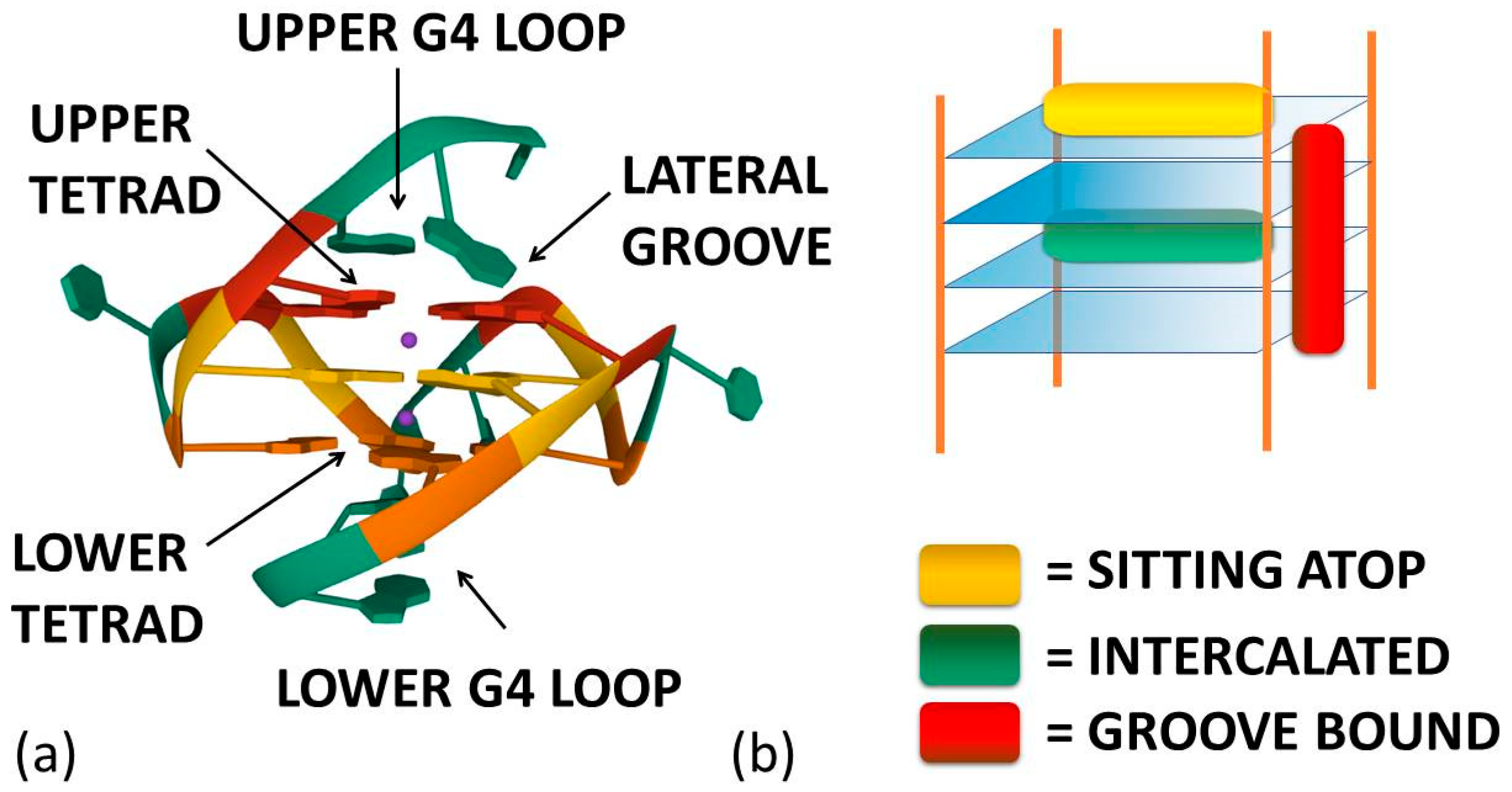
Figure 2. (a) Ligand–G4 interaction sites (the structure is PDB-1XAV, a monomeric parallel-stranded G4 from human c-MYC promoter). (b) Ligand–G4 complex types cartoon: the yellow rectangle refers to a species whose G4 interaction is driven by upper G4 stacking; the green one refers to a species able to intercalate itself between two adjacent tetrads; the red one refers to a molecule that chooses lateral grooves as the preferred binding site.
2. G4 ligands
2.1. Porphyrins
TMpyP4 can bind to hybrid structures, both by the external G-tetrad planes and with the bases of the interconnecting loops; this interaction produces a destabilisation of the hybrid geometry and finally leads to a switch to basket-type antiparallel topologies. However, this occurs under diluted conditions only; the same authors analysed what happens under molecular crowding conditions (40% w/v of PEG200) and found that the parallel structure becomes the majority and is not affected anymore by TMPyP4 binding [41]. Methyl mesoporphyrin IX increases the parallel component of different G4s [42]. A peculiar behaviour, already evidenced in Ma’s paper, deserves to be highlighted also here: porphyrazine can induce antiparallel quadruplex conformations and can be used also along with a complementary ligand to controllably switch quadruplex conformations between parallel and antiparallel topologies (Figure 3) [43]. Note that even more complicated systems can be engineered: N-methyl mesoporphyrin IX (NMM) was utilized to first convert a hybrid G4 into the parallel fold, and then cationic hemicyanine dyes are used to exchange NMM and favour antiparallel or hybrid topologies. Thus, NMM affords a topology that differs from one directly favoured by the hemicyanines and enables the monitoring of the binding by the loss of its fluorescence upon binding [44]. The planar square-based Cu(II) 12-MC-4 metallacrown has some geometrical similarities with porphyrin metal complexes and, similarly to what happens, for instance, in the case of the iron-porphyrin hemin [45][46], induces transformations into the parallel form [47].
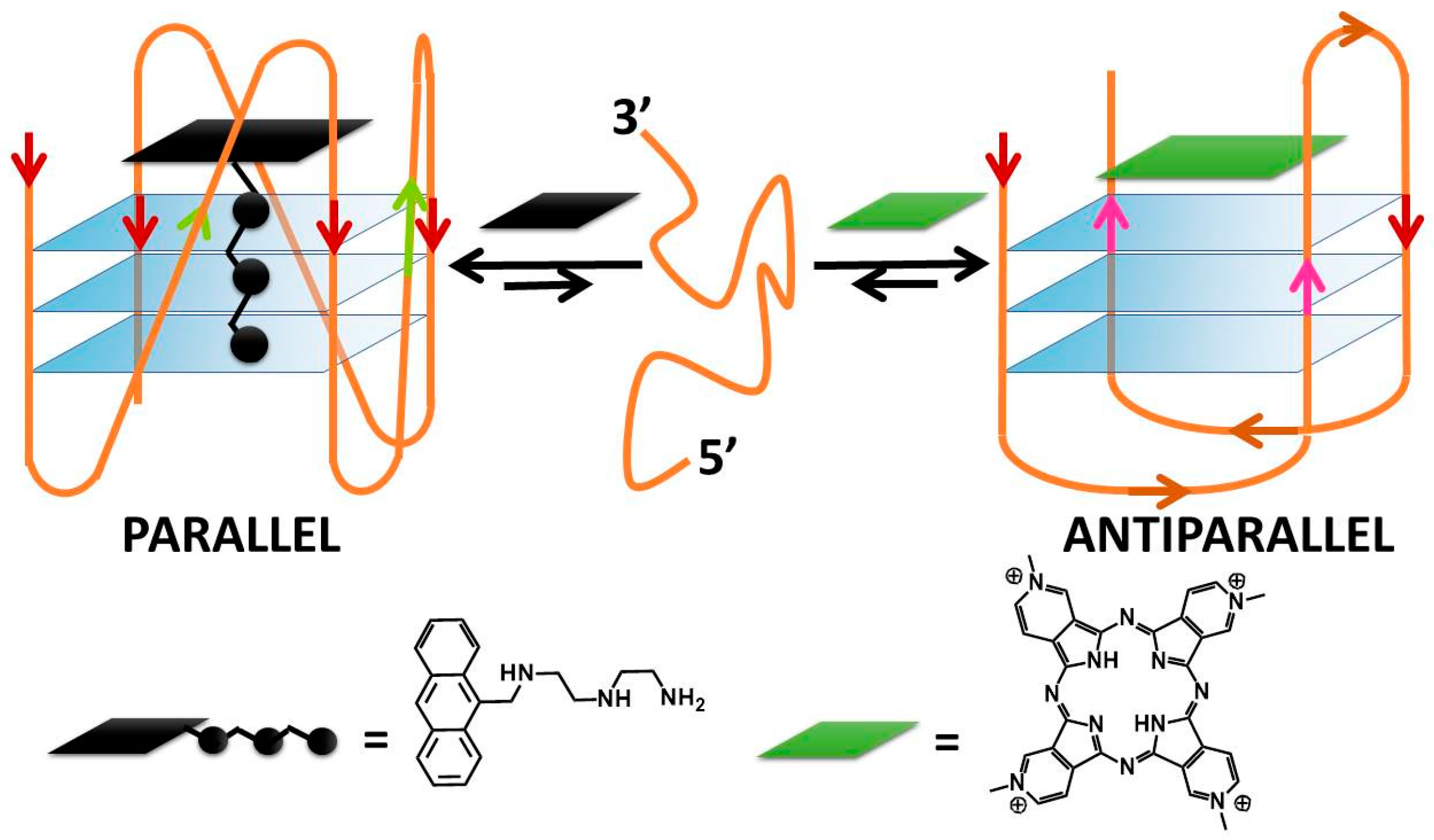
Figure 3. DNA conformational switching according to Rodriguez and co-workers [43].
2.2. Phenanthrolines Derivatives and Their Metal Complexes
A benzophenanthridine derivative (Figure 4a) can switch antiparallel G4s into parallel ones [48]; interestingly, among other similar derivatives, it is the only molecule able to produce this effect on G4 sequences from HIF1α (the subunit of a hypoxia-inducible factor heterodimeric protein) and to consequently interfere with HIF1α binding to a transcriptional factor. The same switching behaviour from antiparallel to parallel is also found for a 9-aminoacridine derivative (Figure 4b) on a G4-forming sequence, which is the one identified in the promoter region of Nrf2 (nuclear factor (erythroid-derived 2)-like 2) [9]. As for metal complexes, ruthenium ones are the more abundantly studied. The Ru–indoloquinoline complex shown in Figure 4c can convert a hybrid telomeric structure into an antiparallel G4 and also destroy the G4 structure in the case of interaction with parallel c-MYC [49]. The hybrid-antiparallel conversion ability is retained when a –OCH3 functionality is added to the indoloquinoline ligand [50]. In this family of ruthenium complexes, the presence of chirality is always an interesting aspect to take into account. The –CN derivative shown in Figure 4d showed a marked enantiospecific behaviour as Λ-[Ru(TAP)2(11-CN-dppz)]2+ (TAP = 1,4,5,8-tetraazaphenanthrene, dppz = dipyridophenazine) changes parallel topologies into antiparallel ones, whereas Δ-[Ru(TAP)2(11-CN-dppz)]2+ is not a switch inducer. This behaviour is analysed in the details thanks to the comparison between crystallographic data, synchrotron radiation circular dichroism (SRCD) and spectroscopic/melting tests, elucidating that the observed behaviour is a consequence of the increased stabilisation of syn-guanosine by the Λ-enantiomer [51].
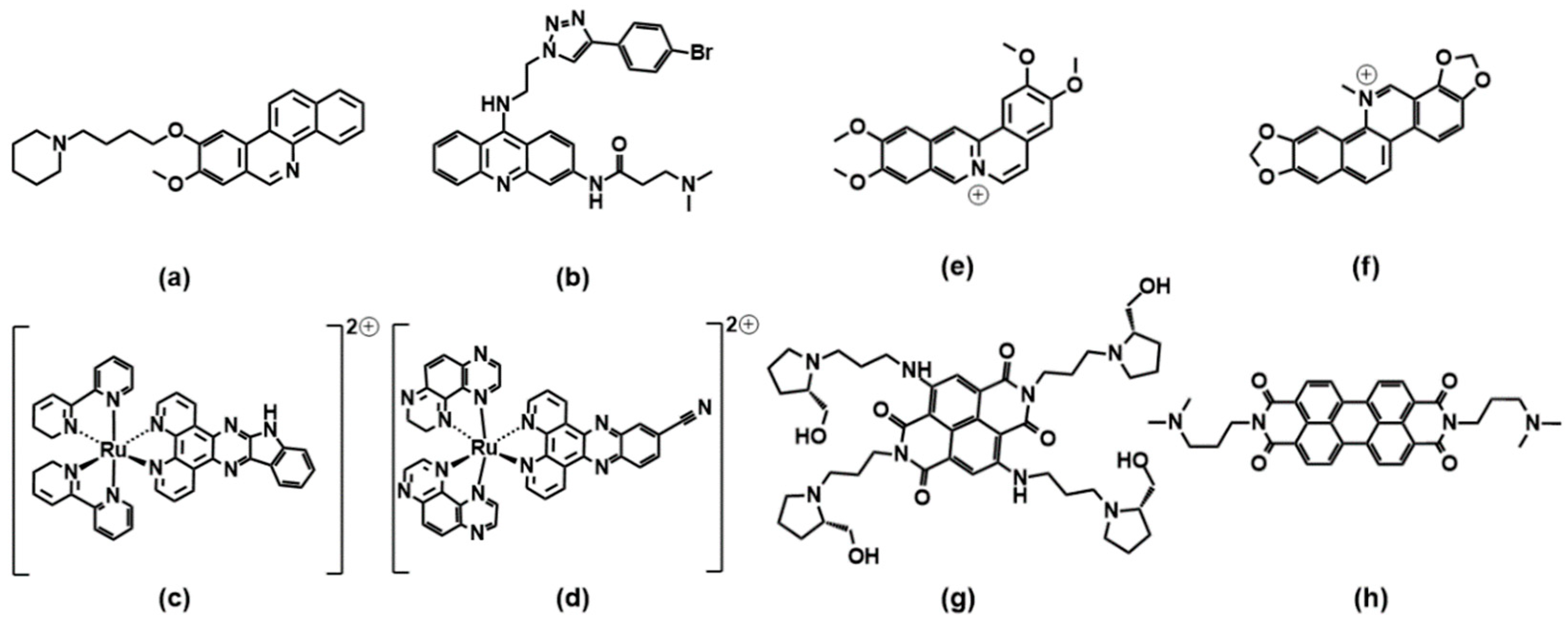
2.3. Pericyclic Compounds
Coralyne (Figure 4e), a known synthetic protoberberine alkaloid, can induce the transformation of hybrid/antiparallel G4 to parallel forms. Detailed circular dichroism, molecular dynamics and 1H/13C/31P NMR analyses produced a picture with coralyne stacking at two upper and lower tetrads, accompanied by the formation of a hydrogen bond; i.e., a highly specific interaction at the molecular level that induces the specific changes in the G4 backbone, which may be the basis for the switch [52]. Similarly, the sanguinarine alkaloid (Figure 4f) can induce the conformational change from hybrid G4 to antiparallel basket-type conformations [53].
The naphthalene diimide (NDI) shown in Figure 4g can induce a switch of G4 morphology from hybrid to parallel; the authors use their spectroscopic data (circular dichroism, NMR and fluorescence tests, including Förster resonance energy transfer experiments) to gain information on a detailed step-by-step reaction mechanism for which they propose the intermediates’ geometries [54]. More extended perylene diimides (PDI) also interfere with the G4 topologies: the molecule shown in Figure 4h is found, in particular by electrospray ionization mass spectrometry, to drive the formation of a parallel G4 form starting from a mixed parallel/antiparallel geometry [55].
[...further information on other compounds may be found at 10.3390/molecules27134165....]
This entry is adapted from the peer-reviewed paper 10.3390/molecules27134165
References
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415.
- Sen, D.; Gilbert, W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, 364–366.
- Kilpatrick, M.W.; Torri, A.; Kang, D.S.; Engler, J.A.; Wells, R.D. Unusual DNA structures in the adenovirus genome. J. Biol. Chem. 1986, 261, 11350–11354.
- Masai, H.; Tanaka, T. G-quadruplex DNA and RNA: Their roles in regulation of DNA replication and other biological functions. Biochem. Biophys. Res. Commun. 2020, 531, 25–38.
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474.
- Bhattacharyya, D.; Arachchilage, G.M.; Basu, S. Metal Cations in G-Quadruplex Folding and Stability. Front. Chem. 2016, 4, 38.
- Ishiguro, A.; Katayama, A.; Ishihama, A. Different recognition modes of G-quadruplex RNA between two ALS/FTLD-linked proteins TDP-43 and FUS. FEBS Lett. 2021, 595, 310–323.
- Ou, T.-M.; Lu, Y.-J.; Tan, J.-H.; Huang, Z.-S.; Wong, K.-Y.; Gu, L.-Q. G-Quadruplexes: Targets in Anticancer Drug Design. Chem. Med. Chem. 2008, 3, 690–713.
- Waller, Z.A.E.; Howell, L.A.; MacDonald, C.J.; O’Connell, M.A.; Searcey, M. Identification and characterisation of a G-quadruplex forming sequence in the promoter region of nuclear factor (erythroid-derived 2)-like 2 (Nrf2). Biochem. Biophys. Res. Commun. 2014, 447, 128–132.
- Ma, Y.; Iida, K.; Nagasawa, K. Topologies of G-quadruplex: Biological functions and regulation by ligands. Biochem. Biophys. Res. Commun. 2020, 531, 3–17.
- Marchand, A.; Gabelica, V. Folding and misfolding pathways of G-quadruplex DNA. Nucleic Acids Res. 2016, 44, 10999–11012.
- Chan, M.S.; Leung, H.M.; Wong, S.W.; Lin, Z.; Gao, Q.; Chang, T.J.H.; Lai, K.W.C.; Lo, P.K. Reversible reconfiguration of high-order DNA nanostructures by employing G-quartet toeholds as adhesive units. Nanoscale 2020, 12, 2464–2471.
- Wright, W.E.; Tesmer, V.M.; Huffman, K.E.; Levene, S.D.; Shay, J.W. Normal human chromosomes have long G-rich telomeric overhangs at one end. Genes Dev. 1997, 11, 2801–2809.
- Hsu, S.-T.D.; Varnai, P.; Bugaut, A.; Reszka, A.P.; Neidle, S.; Balasubramanian, S. A G-Rich Sequence within the c-kit Oncogene Promoter Forms a Parallel G-Quadruplex Having Asymmetric G-Tetrad Dynamics. J. Am. Chem. Soc. 2009, 131, 13399–13409.
- Lim, K.W.; Alberti, P.; Guédin, A.; Lacroix, L.; Riou, J.-F.; Royle, N.J.; Mergny, J.-L.; Phan, A.T. Sequence variant (CTAGGG)n in the human telomere favors a G-quadruplex structure containing a G·C·G·C tetrad. Nucleic Acids Res. 2009, 37, 6239–6248.
- Wang, Z.-F.; Li, M.-H.; Hsu, S.-T.D.; Chang, T.-C. Structural basis of sodium–potassium exchange of a human telomeric DNA quadruplex without topological conversion. Nucleic Acids Res. 2014, 42, 4723–4733.
- Ambrus, A.; Chen, D.; Dai, J.; Bialis, T.; Jones, R.A.; Yang, D. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006, 34, 2723–2735.
- Howard, F.B.; Frazier, J.; Miles, H.T. Stable and metastable forms of poly(G). Biopolymers 1977, 16, 791–809.
- Luu, K.N.; Phan, A.T.; Kuryavyi, V.; Lacroix, L.; Patel, D.J. Structure of the Human Telomere in K+ Solution: An Intramolecular (3 + 1) G-Quadruplex Scaffold. J. Am. Chem. Soc. 2006, 128, 9963–9970.
- Miyoshi, D.; Nakao, A.; Sugimoto, N. Structural transition from antiparallel to parallel G-quadruplex of d(G 4 T 4 G 4) induced by Ca2+. Nucleic Acids Res. 2003, 31, 1156–1163.
- Mergny, J.-L.; Hélène, C. G-quadruplex DNA: A target for drug design. Nat. Med. 1998, 4, 1366–1367.
- Wheelhouse, R.T.; Sun, D.; Han, H.; Han, F.X.; Hurley, L.H. Cationic Porphyrins as Telomerase Inhibitors: the Interaction of Tetra-(N-methyl-4-pyridyl)porphine with Quadruplex DNA. J. Am. Chem. Soc. 1998, 120, 3261–3262.
- Yaku, H.; Fujimoto, T.; Murashima, T.; Miyoshi, D.; Sugimoto, N. Phthalocyanines: A new class of G-quadruplex-ligands with many potential applications. Chem. Commun. 2012, 48, 6203–6216.
- Yurenko, Y.P.; Novotný, J.; Marek, R. Weak Supramolecular Interactions Governing Parallel and Antiparallel DNA Quadruplexes: Insights from Large-Scale Quantum Mechanics Analysis of Experimentally Derived Models. Chem. Eur. J. 2017, 23, 5573–5584.
- Haase, L.; Weisz, K. Locked nucleic acid building blocks as versatile tools for advanced G-quadruplex design. Nucleic Acids Res. 2020, 48, 10555–10566.
- Dai, J.; Carver, M.; Punchihewa, C.; Jones, R.A.; Yang, D. Structure of the Hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: Insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res. 2007, 35, 4927–4940.
- Masiero, S.; Trotta, R.; Pieraccini, S.; De Tito, S.; Perone, R.; Randazzo, A.; Spada, G.P. A non-empirical chromophoric interpretation of CD spectra of DNA G-quadruplex structures. Org. Biomol. Chem. 2010, 8, 2683–2692.
- Poudel, L.; Steinmetz, N.F.; French, R.H.; Parsegian, V.A.; Podgornik, R.; Ching, W.-Y. Implication of the solvent effect, metal ions and topology in the electronic structure and hydrogen bonding of human telomeric G-quadruplex DNA. Phys. Chem. Chem. Phys. 2016, 18, 21573–21585.
- Price, D.A.; Wedamulla, P.; Hill, T.D.; Loth, T.M.; Moran, S.D. The polarization dependence of 2D IR cross-peaks distinguishes parallel-stranded and antiparallel-stranded DNA G-quadruplexes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 267, 120596.
- Georgiades, S.N.; Abd Karim, N.H.; Suntharalingam, K.; Vilar, R. Interaction of Metal Complexes with G-Quadruplex DNA. Angew. Chem. Int. Ed. 2010, 49, 4020–4034.
- Arola, A.; Vilar, R. Stabilisation of G-Quadruplex DNA by Small Molecules. Curr. Top. Med. Chem. 2008, 8, 1405–1415.
- Busto, N.; Calvo, P.; Santolaya, J.; Leal, J.M.; Guédin, A.; Barone, G.; Torroba, T.; Mergny, J.-L.; García, B. Fishing for G-Quadruplexes in Solution with a Perylene Diimide Derivative Labeled with Biotins. Chem. Eur. J. 2018, 24, 11292–11296.
- Neidle, S. Quadruplex nucleic acids as targets for anticancer therapeutics. Nat. Rev. Chem. 2017, 1, 41.
- Monchaud, D.; Granzhan, A.; Saettel, N.; Guédin, A.; Mergny, J.-L.; Teulade-Fichou, M.-P. “One Ring to Bind Them All”—Part I: The Efficiency of the Macrocyclic Scaffold for G-Quadruplex DNA Recognition. J. Nucleic Acids 2010, 2010, 525862.
- Pérez-Arnaiz, C.; Busto, N.; Santolaya, J.; Leal, J.M.; Barone, G.; García, B. Kinetic evidence for interaction of TMPyP4 with two different G-quadruplex conformations of human telomeric DNA. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 522–531.
- Cao, Q.; Li, Y.; Freisinger, E.; Qin, P.Z.; Sigel, R.K.O.; Mao, Z.-W. G-quadruplex DNA targeted metal complexes acting as potential anticancer drugs. Inorg. Chem. Front. 2017, 4, 10–32.
- Reed, J.E.; Arnal, A.A.; Neidle, S.; Vilar, R. Stabilization of G-Quadruplex DNA and Inhibition of Telomerase Activity by Square-Planar Nickel(II) Complexes. J. Am. Chem. Soc. 2006, 128, 5992–5993.
- Monchaud, D.; Teulade-Fichou, M.-P. A hitchhiker’s guide to G-quadruplex ligands. Org. Biomol. Chem. 2008, 6, 627–636.
- Rajput, C.; Rutkaite, R.; Swanson, L.; Haq, I.; Thomas, J.A. Dinuclear Monointercalating RuII Complexes That Display High Affinity Binding to Duplex and Quadruplex DNA. Chem. Eur. J. 2006, 12, 4611–4619.
- Han, H.; Langley, D.R.; Rangan, A.; Hurley, L.H. Selective Interactions of Cationic Porphyrins with G-Quadruplex Structures. J. Am. Chem. Soc. 2001, 123, 8902–8913.
- Martino, L.; Pagano, B.; Fotticchia, I.; Neidle, S.; Giancola, C. Shedding Light on the Interaction between TMPyP4 and Human Telomeric Quadruplexes. J. Phys. Chem. B 2009, 113, 14779–14786.
- Nicoludis, J.M.; Barrett, S.P.; Mergny, J.-L.; Yatsunyk, L.A. Interaction of human telomeric DNA with N- methyl mesoporphyrin IX. Nucleic Acids Res. 2012, 40, 5432–5447.
- Rodriguez, R.; Pantoş, G.D.; Gonçalves, D.P.N.; Sanders, J.K.M.; Balasubramanian, S. Ligand-driven G-quadruplex conformational switching by using an unusual mode of interaction. Angew. Chem. Int. Ed. Engl. 2007, 46, 5405–5407.
- Deore, P.S.; Gray, M.D.; Chung, A.J.; Manderville, R.A. Ligand-Induced G-Quadruplex Polymorphism: A DNA Nanodevice for Label-Free Aptasensor Platforms. J. Am. Chem. Soc. 2019, 141, 14288–14297.
- Brooks, T.A.; Hurley, L.H. The role of supercoiling in transcriptional control of MYC and its importance in molecular therapeutics. Nat. Rev. Cancer 2009, 9, 849–861.
- Kong, D.-M.; Yang, W.; Wu, J.; Li, C.-X.; Shen, H.-X. Structure–function study of peroxidase-like G-quadruplex-hemin complexes. Analyst 2010, 135, 321–326.
- Rajczak, E.; Juskowiak, B. Conformational rearrangements of G-quadruplex topology promoted by Cu(II) 12-MCCu(II)PyrAcHA-4 metallacrown. Int. J. Biol. Macromol. 2020, 156, 1258–1269.
- Chen, H.; Long, H.; Cui, X.; Zhou, J.; Xu, M.; Yuan, G. Exploring the Formation and Recognition of an Important G-Quadruplex in a HIF1α Promoter and Its Transcriptional Inhibition by a Benzophenanthridine Derivative. J. Am. Chem. Soc. 2014, 136, 2583–2591.
- Yu, H.-J.; Zhao, Y.; Mo, W.-J.; Hao, Z.-F.; Yu, L. Ru-indoloquinoline complex as a selective and effective human telomeric G-quadruplex binder. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2014, 132, 84–90.
- Yu, H.-J.; Yu, L.; Hao, Z.-F.; Zhao, Y. Interactions of ruthenium complexes containing indoloquinoline moiety with human telomeric G-quadruplex DNA. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2014, 124, 187–193.
- McQuaid, K.; Abell, H.; Gurung, S.P.; Allan, D.R.; Winter, G.; Sorensen, T.; Cardin, D.J.; Brazier, J.A.; Cardin, C.J.; Hall, J.P. Structural Studies Reveal Enantiospecific Recognition of a DNA G-Quadruplex by a Ruthenium Polypyridyl Complex. Angew. Chem. Int. Ed. 2019, 58, 9881–9885.
- Padmapriya, K.; Barthwal, R. NMR based structural studies decipher stacking of the alkaloid coralyne to terminal guanines at two different sites in parallel G-quadruplex DNA, 4 and 4. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2017, 1861, 37–48.
- Pradhan, S.K.; Dasgupta, D.; Basu, G. Human telomere d undergoes a conformational transition to the Na+-form upon binding with sanguinarine in presence of K+. Biochem. Biophys. Res. Commun. 2011, 404, 139–142.
- Hao, X.; Wang, C.; Wang, Y.; Li, C.; Hou, J.; Zhang, F.; Kang, C.; Gao, L. Topological conversion of human telomeric G-quadruplexes from hybrid to parallel form induced by naphthalene diimide ligands. Int. J. Biol. Macromol. 2021, 167, 1048–1058.
- Zhou, J.; Yuan, G. Specific Recognition of Human Telomeric G-Quadruplex DNA with Small Molecules and the Conformational Analysis by ESI Mass Spectrometry and Circular Dichroism Spectropolarimetry. Chemistry 2007, 13, 5018–5023.
This entry is offline, you can click here to edit this entry!