The advancement of new therapies, including targeted therapies and immunotherapies, has improved the survival of non-small-cell lung cancer (NSCLC) patients in the last decade. Some NSCLC patients still do not benefit from therapies or encounter progressive disease during the course of treatment because they have intrinsic resistance, acquired resistance, or lack a targetable driver mutation. More investigations on the molecular biology of NSCLC are needed to find useful biomarkers for current therapies and to develop novel therapeutic strategies. Src is a non-receptor tyrosine kinase protein that interacts with cell surface growth factor receptors and the intracellular signaling pathway to maintain cell survival tumorigenesis in NSCLC. The Yes-associated protein (YAP) is one of the main effectors of the Hippo pathway and has been identified as a promoter of drug resistance, cancer progression, and metastasis in NSCLC. Here, we review studies that have investigated the activation of YAP as mediated by Src kinases and demonstrate that Src regulates YAP through three main mechanisms: (1) direct phosphorylation; (2) the activation of pathways repressing Hippo kinases; and (3) Hippo-independent mechanisms. Further work should focus on the efficacy of Src inhibitors in inhibiting YAP activity in NSCLC. In addition, future efforts toward developing potentially reasonable combinations of therapy targeting the Src–YAP axis using other therapies, including targeted therapies and/or immunotherapies, are warranted.
- proto-oncogene tyrosine-protein kinase Src
- yes-associated protein (YAP)
- Hippo pathway
- non-small cell lung cancer (NSCLC)
- tyrosine-kinase inhibitor (TKI)
- YES1
- dasatinib
1. Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide, and most patients are diagnosed at an advanced stage of disease [1][2]. Non-small-cell lung cancer (NSCLC) accounts for 85% of lung cancer cases, according to the classification of histopathology [3][4]. Chemotherapy is used as the standard treatment for most patients with advanced disease, but its effect is limited because of unsatisfactory efficacy and side effects [5]. In the last decade, new therapies that have been developed for advanced NSCLC treatment, including targeted therapy and immunotherapy, have shown better clinical effects in terms of the objective response rate and prolonging survival in comparison with conventional chemotherapy [6][7]. Several oncogenic drivers of NSCLC, such as the epidermal growth factor receptor (EGFR) and BRAF mutations, as well as anaplastic lymphoma kinase (ALK) and ROS-1 rearrangements, have been uncovered and studied. Drugs targeting these oncogenic drivers have been developed, and most of them have been approved by the US Food and Drug Administration (FDA) for the clinical treatment of advanced NSCLC patients harboring oncogenic-driven mutations [6,8,9]. Currently, targeted therapies, including those based on the use of EGFR-tyrosine kinase inhibitors (TKIs), BRAF inhibitors, and ALK inhibitors, have demonstrated promising efficacy, with a 60–80% response rate and 9–30 months of progression-free survival in treating advanced NSCLC with relevant driver mutations [8][9]. Immunotherapy using anti-PD-1/PD-L1 immune checkpoint inhibitors has been developed as a new therapy for metastatic NSCLC that has demonstrated efficacy in numerous clinical trials in which a 15–45% objective response rate was observed in addition to prolonged overall survival [10][11][12]. However, some NSCLC patients still do not have targetable driver mutations and do not benefit from immunotherapy [13]. In addition, acquired resistance to targeted therapies and immunotherapy occur after a period of treatment in most NSCLC patients who had an initial response to the treatment [13]. Acquired resistance to multiple treatment modalities limits the five-year survival rate of NSCLC patients (~16%) [13]. Therefore, further investigation on the molecular biology of NSCLC is needed for the development of novel therapeutic strategies and the determination of useful biomarkers for treatment selection.
Src (also known as proto-oncogene tyrosine protein kinase or c-Src) is a non-receptor tyrosine kinase protein that has been reported to promote tumorigenesis and metastatic progression in various cancers [14][15]. Src has also been identified in human NSCLC and has been reported to interact with several growth factor receptors including EGFR, human epidermal growth factor receptor 2 (HER2), hepatocyte growth factor receptor (HGFR; also known as c-MET), platelet-derived growth factor receptor (PDGFR), insulin-like growth factor receptor (IGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor (VEGFR), and focal adhesion kinase (FAK) [14][15]. Yes-associated protein (YAP) is a key mediator of the Hippo signaling pathway and has been identified as an important oncoprotein in human NSCLC [16][17]. YAP has been reported to interact with growth factor receptors and their downstream signaling pathways to promote drug resistance, cancer progression, and metastasis in NSCLC [18][19].
Here, we review the crosstalk between Src family kinases and the Hippo/YAP signaling pathway and look for new therapeutic targets in human NSCLC for future study.
2. Src in NSCLC
Src is a non-receptor tyrosine kinase protein that consists of the SH1 (tyrosine kinase) SH2, SH3, and SH4 (unique) domains; the SH3–SH2 connector; the SH2–kinase linker; a C-terminal tail regulatory region; and two tyrosine sites (Tyr416 and Tyr527) (Figure 1A.) [20]. The tyrosine site Tyr416 is located in a kinase domain (SH1), while the other tyrosine site, Tyr527, is located in the C-terminal region [20]. The intramolecular interactions among the domains and phosphorylation of the two tyrosine sites are crucial for the regulation of Src. Src is usually present in an inactive form when phosphorylated Tyr527 binds to the SH2 domain and the SH2–kinase linker binds to the SH3 domain. This conformation protects the catalytic pocket of Tyr 416 in the kinase domain (SH1) from inappropriate phosphorylation. The dephosphorylation of Ty527 causes a conformational change that unlocks the catalytic pocket of Tyr 416 and leads to subsequent activation of Src by the intramolecular autophosphorylation of Tyr416 [20][21] (Figure 1B).
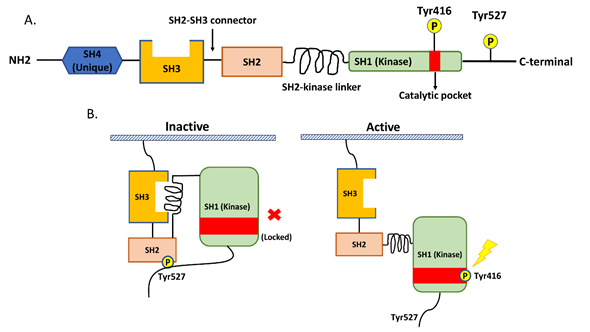
Figure 1. The structure of Src and the regulation of its kinase activity. (A) Src consists of the SH1 (tyrosine kinase), SH2, SH3, and SH4 (unique) domains; the SH3–SH2 connector; the SH2–kinase linker; a C-terminal tail regulatory region; and two tyrosine sites (Tyr416 and Tyr527). (B) The regulation of Src activity depends on the phosphorylation of two tyrosine sites and intramolecular interactions among the domains. Normally, phosphorylated Tyr527 binds to the SH2 domain and the SH2–kinase linker binds to the SH3 domain, and these binding events result in the protection of the catalytic pocket of Tyr 416 in the kinase domain (SH1) from inappropriate phosphorylation. The dephosphorylation of Ty527 results in conformational change, unlocking the catalytic pocket of Tyr 416 and subsequently activating Src through the intramolecular autophosphorylation of Tyr416.
Src protein kinase is expressed in the cells of normal tissues including the brain, bone, lungs, leukocytes, and platelets. Src is involved in the transduction of signals from the cell surface receptors and plays an important role in regulating cell growth, differentiation, and survival [22][23][24]. Regarding the regulation of intracellular signaling transduction, Src has been identified as an important oncoprotein that promotes cancer progression, invasion, metastasis, and drug resistance in various cancers, including colorectal cancer, breast cancer, pancreatic cancer, gastric cancer, and lung cancer [25][26][27]. The overexpression and/or hyperactivation of Src have been reported in human cancers and lead to the upregulation of various receptors of tyrosine kinases (RTKs), including EGFR, HER2, c-MET, PDGFR, IGFR, FGFR, and VEGFR . In addition, Src interacts with other transmembrane receptors, such as integrins/Focal adhesion kinase (FAK) and G-protein-coupled receptors (GPCRs), to upregulate their downstream signaling [28][29][30]. Regarding human NSCLC, previous studies have shown that the overexpression of Src is associated with poor prognosis and the promotion of migration, invasion, metastasis, and drug resistance [31][32][33][34]. EGFR kinase domain mutations, such as L858R and exon 19 deletion, are the most frequent oncogenic-driven mutations in human NSCLC (5–15% in Caucasians and 40–55% in East Asians), and EGFR-tyrosine kinase inhibitors (TKIs) have been developed as an effective therapy for advanced EGFR-mutated NSCLC [35][36][37]. EGFR kinase domain mutations activate downstream signaling pathways, including the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK), phosphatidylinositol 3‑kinase (PI3K)/Akt/mTOR, and interleukin 6 (IL-6)/Janus kinase (JAK)/signal transducer, and the activator of transcription 3 (STAT3) signaling pathways [34]. Several previous studies have provided evidence that Src activates the three downstream signaling pathways (MAPK/ERK, PI3K/Akt/mTOR, and IL-6/JAK/STAT3), thus promoting resistance to EGFR-TKIs in NSCLC [38][39]. A recent study found that the Src family kinase YES1 (v-YES-1 Yamaguchi sarcoma viral oncogene homolog 1) was amplified in NSCLC patients harboring EGFR mutations or echinoderm microtubule-associated protein-like 4 (EML4)-anaplastic lymphoma kinase (ALK) fusion who had acquired resistance to EGFR or ALK inhibitors [40]. Therefore, the amplification of the Src family kinase YES1 was identified as a mechanism for acquired resistance to EGFR and ALK inhibitors. In a study by Fan et al., it was demonstrated that the forced overexpression of YES1 promotes resistance to EGFR-TKIs in the human NSCLC cell line PC9, and the inhibition of Src re-enhances the cytotoxicity of EGFR-TKIs [40]. Two previous studies also demonstrated that inhibiting Src family kinase enhances the antitumor effect of EGFR-TKIs on EGFR-mutated NSCLC cells [41]. Studies about Src-altered resistance to targeted therapy in NSCLC are summarized in Table 1.
Table 1. Src-altered resistance to targeted therapy in non-small-cell lung cancer (NSCLC).
|
Genomic Alternation of Src |
Primary Mutation of NSCLC |
Resistance to Target Therapy (Primary/Secondary) |
Targeted Therapy Resistance |
Type of Study |
Reference |
|
Overexpression of SHP2 |
EGFR mutation L858R/exon 19 deletion /G719X/L861Q |
Primary |
Erlotinib, Gefitinib, Afatinib |
Clinical analysis |
[39] |
|
YES1 amplification |
EGFR mutation L858R/exon 19 deletion |
Secondary |
Erlotinib, Afatinib |
Clinical analysis |
[37] |
|
YES1 amplification |
EGFR mutation Exon 19 deletion/ L858R+T790M |
Secondary |
Osimertinib |
Preclinical study |
[38] |
|
YES1 amplification |
EGFR mutation L858R/exon 19 deletion ALK fusion EML4-ALK fusion/ HIP1-ALK fusion |
Secondary |
Erlotinib, Afatinib, Crizotinib, Ceritinib |
Clinical analysis |
[40] |
ALK: anaplastic lymphoma kinase; EGFR: epidermal growth factor receptor; EML4: echinoderm microtubule-associated protein-like 4.
G-protein-coupled receptors (GPCRs) constitute a large family of diverse receptors that play crucial roles in the function of normal tissues through the activation of intracellular signaling pathways in response to specific extracellular signals. GPCRs have been found to play an important role in tumorigenesis and cancer progression and have been reported to be overexpressed in human NSCLC [42][43]. Therefore, therapeutic approaches targeting GPCRs in metastatic NSCLC have been developed, and their efficacy is currently under investigation [44][45]. Previous studies have shown that the downstream signaling transduction of GPCRs depends on the phosphorylation of Src kinase in some manner [46][47]. Another two previous studies demonstrated that the crosstalk between GPCRs and EGFR is bridged by Src kinase in human NSCLC cells. They found that some GPCRs, such as formyl peptide receptor-like 1 (FPRL1) and cholecystokinin (CCK) receptors, are present in human NSCLC cells and transactivate EGFR downstream signaling pathways, including MAPK and JAK/STAT3, to promote cell proliferation through Src kinase. In addition, both studies showed that the inhibition of Src blocks the transactivation of EGFR induced by the GPCRs [48][49]. The Rho/Rho-associated protein kinase (ROCK) pathway is a downstream signaling pathway that is regulated by Src and is involved in the promotion of cancer invasion and metastasis [50]. The co-expression of Rho and Src in human NSCLC has been reported, and therapy based on the targeting of Rho/ROCK in metastatic NSCLC has been investigated in several studies [51][52][53]. Onodera et al. showed that the concurrent inhibition of Src and Rho/ROCK has a synergistic effect in suppressing NSCLC cell growth [53].
The Src kinase protein plays an important role in mediating signaling transduction between transmembrane cell surface receptors and downstream intracellular pathways. The gene amplification of YES1 (one of the Src family kinases) has been found to partly impact the clinical prognosis of stage I or II NSCLC in previous studies [54]. To date, YES1 is the only SRC family kinase member that has been found to be regulated by gene amplification, and a high correlation has been demonstrated between the gene copy number and mRNA expression in human NSCLC [55]. Further, the experimental findings of a recent study conducted by Garmendia et al. suggest that molecular alterations of YES1 could be used as a prognostic biomarker and therapeutic target in human NSCLC [56]. First, they showed that NSCLC patients with high YES1 expression had significantly shorter overall survival times than those with low YES1 expression. Second, YES1 overexpression significantly increased NSCLC cell proliferation in vitro and induced metastatic spread in preclinical mouse models. Third, the knockdown of the YES1 gene decreased NSCLC cell proliferation, invasion ability, and tumor growth in vivo. In addition, the antitumor effect of the Src inhibitor dasatinib was investigated. It was found that dasatinib has significantly higher cytotoxicity towards NSCLC cells with high rather than low YES1 expression. Furthermore, dasatinib treatment was found to significantly inhibit tumor growth in a patient-derived xenograft (PDX) model with high YES1 expression. In these PDX models, dasatinib treatment did not affect tumor growth in patients with low YES1 expression. The study suggests that selected advanced NSCLC patients with high YES1 expression or genetic amplification may benefit from dasatinib treatment [56].
The interactions among transmembrane cell surface receptors, Src kinase, and downstream signaling pathways are summarized in Figure 2.
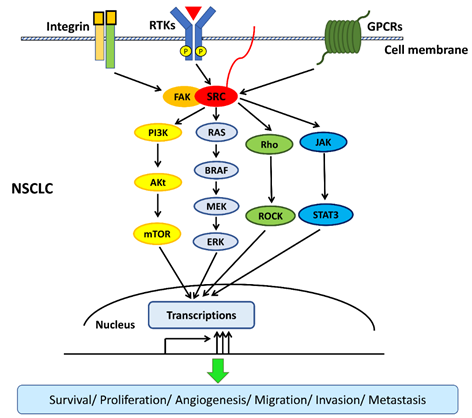
Figure 2. Src interacts with transmembrane cell surface receptors and mediates the transduction of intracellular downstream signaling pathways. Src interacts with transmembrane cell surface receptors, including the integrin/focal adhesion kinase (FAK), the receptor of tyrosine kinases (RTKs), and G-protein-coupled receptors (GPCRs), and then activates downstream signaling pathways. Src activates downstream signaling pathways, including mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK), phosphatidylinositol 3‑kinase (PI3K)/Akt/mTOR, interleukin 6 (IL-6)/Janus kinase (JAK)/signal transducer and the activator of transcription 3 (STAT3), and Rho/Rho-associated protein kinase (ROCK) pathways to promote survival, proliferation, angiogenesis, migration, invasion, and metastasis in non-small-cell lung cancer (NSCLC).
This entry is adapted from the peer-reviewed paper 10.3390/cancers12061361
References
- McIntyre, A.; Ganti, A.K. Lung cancer-A global perspective. J. Surg. Oncol. 2017, 115, 550–554.
- Cao, M.; Chen, W. Epidemiology of lung cancer in China. Thorac. Cancer 2019, 10, 3–7.
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009.
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546.
- Lazzari, C.; Bulotta, A.; Ducceschi, M.; Viganò,, M.G.; Brioschi, E.; Corti, F.; Gianni, L.; Gregorc, V. Historical Evolution of Second-Line Therapy in Non-small-cell Lung Cancer. Front Med. 2017, 4, 4.
- Saber, A.; van der Wekken, A.; Hiltermann, T.J.; Kok, K.; Van den Berg, A.; Groen, H. Genomic aberrations guiding treatment of non-small-cell lung cancer patients. Cancer Treat. Commun. 2015, 4, 23–33.
- Ogunleyea, F.; Blankenshipa, L.; Millisorb, V.; Anderson, J.; Jaiyesimi, I. Programmed cell death-1/Programmed cell death ligand-1(PD-1/PD-L1) inhibitors, heralding a new era of immunotherapy in the management of advanced Non-small-cell Lung Cancer (NSCLC). Cancer Treat. Res. Commun. 2017, 12, 6–13.
- Singal, G.; Miller, P.G.; Agarwala, V.; Li, G.; Kaushik, G.; Backenroth, D.; Gossai, A.; Frampton, G.M.; Torres, A.Z.; Lehnert, E.M.; et al. Association of Patient Characteristics and Tumor Genomics With Clinical Outcomes Among Patients With Non-Small Cell Lung Cancer Using a Clinicogenomic Database. JAMA 2019, 321, 1391–1399.
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small-cell lung cancer. Nature 2018, 553, 446–454.
- Hsu, P.C.; Yang, C.T.; Jablons, D.M.; You, L. The Role of Yes-Associated Protein (YAP) in Regulating Programmed Death-Ligand 1 (PD-L1) in Thoracic Cancer. Biomedicines 2018, 6, 114.
- Perrotta, F.; Rocco, D.; Vitiello, F.; De Palma, R.; Guerra, G.; De Luca, A.; Navani, N.; Bianco, A. Immune Checkpoint Blockade for Advanced NSCLC: A New Landscape for Elderly Patients. Int. J. Mol. Sci. 2019, 20, 2258.
- Hsu, P.C.; Jablons, D.M.; Yang, C.T.; You, L. Epidermal Growth Factor Receptor (EGFR) Pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-small-cell Lung Cancer (NSCLC). Int. J. Mol. Sci. 2019, 20, 3821.
- Hanna, N.H.; Schneider, B.J.; Temin, S.; Baker, S., Jr.; Brahmer, J.; Ellis, P.M.; Gaspar, L.E.; Haddad, R.Y.; Hesketh, P.J.; Jain, D.; et al. Therapy for Stage IV Non-Small-Cell Lung Cancer Without Driver Alterations: ASCO and OH (CCO) Joint Guideline Update. J Clin Oncol. 2020;38(14):1608–1632.
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25.
- Liu, W.; Kovacevic, Z.; Peng, Z.; Jin, R.; Wang, P.; Yue, F.; Zheng, M.; Huang, M.L.; Jansson, P.J.; Richardson, V.; et al. The molecular effect of metastasis suppressors on Src signaling and tumorigenesis: New therapeutic targets. Oncotarget 2015, 6, 35522–35541.
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79.
- Guo, L.; Teng, L. YAP/TAZ for cancer therapy: Opportunities and challenges (review). Int. J. Oncol. 2015, 46, 1444–1452.
- Janse van Rensburg, H.J.; Yang, X. The roles of the Hippo pathway in cancer metastasis. Cell Signal. 2016, 28, 1761–1772.
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658.
- Boggon, T.J.; Eck, M.J. Structure and regulation of Src family kinases. Oncogene 2004, 23, 7918–7927.
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909.
- Martin, G.S. The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2001, 2, 467–475.
- Okutani, D.; Lodyga, M.; Han, B.; Liu, M. Src Protein Tyrosine Kinase Family and Acute Inflammatory Responses. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L129–L141.
- Li, L.F.; Kao, K.C.; Liu, Y.Y.; Lin, C.W.; Chen, N.H.; Lee, C.S.; Wang, C.W.; Yang, C.T. Nintedanib reduces ventilation-augmented bleomycin-induced epithelial-mesenchymal transition and lung fibrosis through suppression of the Src pathway. J. Cell Mol. Med. 2017, 21, 2937–2949.
- Montero, J.C.; Seoane S, Ocaña A, Pandiella, A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: Possible combinations in solid tumors. Clin. Cancer Res. 2011, 17, 5546–5552.
- Liu, C.J.; Kuo, F.C.; Wang, C.L.; Kuo, C.H.; Wang, S.S.; Chen, C.Y.; Huang, Y.B.; Cheng, K.H.; Yokoyama, K.K.; Chen, C.L.; et al. Suppression of IL-8-Src signalling axis by 17β-estradiol inhibits human mesenchymal stem cells-mediated gastric cancer invasion. J. Cell Mol. Med. 2016, 20, 962–972.
- Zhang, J.; Zhang, F.; Niu, R. Functions of Shp2 in cancer. J. Cell Mol. Med. 2015, 19, 2075–2083.
- New, D.C.; Wong, Y.H. Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J. Mol. Signal. 2007, 2, 2.
- Nagathihalli, N.S.; Merchant, N.B. Src-mediated regulation of E-cadherin and EMT in pancreatic cancer. Front Biosci. 2012, 17, 2059–2069.
- Gargalionis, A.N.; Karamouzis, M.V.; Papavassiliou, A.G. The molecular rationale of Src inhibition in colorectal carcinomas. Int. J. Cancer 2014, 134, 2019–2029.
- He, P.; Wu, W.; Wang, H.; Liao, K.; Zhang, W.; Xiong, G.; Wu, F.; Meng, G.; Yang, K. Co-expression of Rho guanine nucleotide exchange factor 5 and Src associates with poor prognosis of patients with resected non-small-cell lung cancer. Oncol. Rep. 2013, 30, 2864–2870.
- Kuranami, S.; Yokobori, T.; Mogi, A.; Altan, B.; Yajima, T.; Onozato, R.; Azuma, Y.; Iijima, M.; Kosaka, T.; Kuwano, H. Src kinase-associated phosphoprotein2 expression is associated with poor prognosis in non-small-cell lung cancer. Anticancer Res. 2015, 35, 2411–2415.
- Gu, Z.; Fang, X.; Li, C.; Chen, C.; Liang, G.; Zheng, X.; Fan, Q. Increased PTPRA expression leads to poor prognosis through c-Src activation and G1 phase progression in squamous cell lung cancer. Int. J. Oncol. 2017, 51, 489–497.
- Liu, W.; Liang, Y.; Chan, Q.; Jiang, L.; Dong, J. CX3CL1 promotes lung cancer cell migration and invasion via the Src/focal adhesion kinase signaling pathway. Oncol. Rep. 2019, 41, 1911–1917.
- Paz-Ares, L.; Soulières, D.; Moecks, J.; Bara, I.; Mok, T.; Klughammer, B. Pooled analysis of clinical outcome for EGFR TKI-treated patients with EGFR mutation-positive NSCLC. J. Cell. Mol. Med. 2014, 18, 1519–1539.
- Li, K.; Yang, M.; Liang, N.; Li, S. Determining EGFR-TKI sensitivity of G719X and other uncommon EGFR mutations in non-small-cell lung cancer: Perplexity and solution (Review). Oncol. Rep. 2017, 37, 1347–1358.
- Yu, H.A.; Suzawa, K.; Jordan, E.; Zehir, A.; Ni, A.; Kim, R.; Kris, M.G.; Hellmann, M.D.; Li, B.T.; Somwar, R.; et al. Concurrent Alterations in EGFR-Mutant Lung Cancers Associated with Resistance to EGFR Kinase Inhibitors and Characterization of MTOR as a Mediator of Resistance. Clin. Cancer Res. 2018, 24, 3108–3118.
- Ichihara, E.; Westover, D.; Meador, C.B.; Yan, Y.; Bauer, J.A.; Lu, P.; Ye, F.; Kulick, A.; de Stanchina, E.; McEwen, R.; et al. SFK/FAK Signaling Attenuates Osimertinib Efficacy in Both Drug-Sensitive and Drug-Resistant Models of EGFR-Mutant Lung Cancer. Cancer Res. 2017, 77, 2990–3000.
- Karachaliou, N.; Cardona, A.F.; Bracht, J.W.P.; Aldeguer, E.; Drozdowskyj, A.; Fernandez-Bruno, M.; Chaib, I.; Berenguer, J.; Santarpia, M.; et al. Integrin-linked kinase (ILK) and src homology 2 domain-containing phosphatase 2 (SHP2): Novel targets in EGFR-mutation positive non-small-cell lung cancer (NSCLC). EBioMedicine 2019, 39, 207–214.
- Fan, P.D.; Narzisi, G.; Jayaprakash, A.D.; Venturini, E.; Robine, N.; Smibert, P.; Germer, S.; Yu, H.A.; Jordan, E.J.; Paik, P.K.; et al. YES1 amplification is a mechanism of acquired resistance to EGFR inhibitors identified by transposon mutagenesis and clinical genomics. Proc. Natl. Acad. Sci. USA. 2018, 115, E6030–E6038.
- Yuan, M.; Xu, L.F.; Zhang, J.; Kong, S.Y.; Wu, M.; Lao, Y.Z.; Zhou, H.; Zhang, L.; Xu, H. SRC and MEK Co-inhibition Synergistically Enhances the Anti-tumor Effect in Both Non-small-cell Lung Cancer (NSCLC) and Erlotinib-Resistant NSCLC. Front Oncol. 2019, 9, 586.
- Khan, M.; Huang, T.; Lin, C.Y.; Wu, J.; Fan, B.M.; Bian, Z.X. Exploiting cancer's phenotypic guise against itself: Targeting ectopically expressed peptide G-protein coupled receptors for lung cancer therapy. Oncotarget 2017, 8, 104615–104637.
- Jiang, X.; Xu, X.; Wu, M.; Guan, Z.; Su, X.; Chen, S.; Wang, H.; Teng, L. GPRC5A: An Emerging Biomarker in Human Cancer. Biomed. Res. Int. 2018, 2018, 1823726.
- Cisowski, J.; O’Callaghan, K.; Kuliopulos, A.; Yang, J.; Nguyen, N.; Deng, Q.; Agarwal, A. Targeting protease-activated receptor-1 with cell-penetrating pepducins in lung cancer. Am. J. Pathol. 2011, 179, 513–523.
- Covic, L.; Kuliopulos, A. Protease-Activated Receptor 1 as Therapeutic Target in Breast, Lung, and Ovarian Cancer: Pepducin Approach. Int. J. Mol. Sci. 2018, 19, 2237.
- Hauser, M.A.; Schaeuble, K.; Kindinger, I.; Impellizzieri, D.; Krueger, W.A.; Hauck, C.R.; Boyman, O.; Legler, D.F. Inflammation-Induced CCR7 Oligomers Form Scaffolds to Integrate Distinct Signaling Pathways for Efficient Cell Migration. Immunity 2016, 44, 59–72.
- Förster, R.; Worbs, T.; Werth, K. A 4-midable Connection: CCR7 Tetramers Link GPCR to Src Kinase Signaling. Immunity 2016, 44, 9–11.
- Cattaneo, F.; Iaccio, A.; Guerra, G.; Montagnani, S.; Ammendola, R. NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic. Biol. Med. 2011, 51, 1126–1136.
- Moody, T.W.; Nuche-Berenguer, B.; Moreno, P.; Jensen, R.T. CI-988 Inhibits EGFR Transactivation and Proliferation Caused by Addition of CCK/Gastrin to Lung Cancer Cells. J. Mol. Neurosci. 2015, 56, 663–672.
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401.
- Tsai, Y.M.; Yang, C.J.; Hsu, Y.L.; Wu, L.Y.; Tsai, Y.C.; Hung, J.Y.; Lien, C.T.; Huang, M.S.; Kuo, P.L. Glabridin inhibits migration, invasion, and angiogenesis of human non-small-cell lung cancer A549 cells by inhibiting the FAK/rho signaling pathway. Integr. Cancer Ther. 2011, 10, 341–349.
- Umelo, I.A.; Wever, O.D.; Kronenberger, P.; Noor, A.; Teugels, E.; Chen, G.; Bracke, M.; Grève, J.D. Combined inhibition of rho-associated protein kinase and EGFR suppresses the invasive phenotype in EGFR-dependent lung cancer cells. Lung Cancer 2015, 90, 167–174.
- Onodera, K.; Sakurada, A.; Notsuda, H.; Watanabe, T.; Matsuda, Y.; Noda, M.; Endo, C.; Okada, Y. Growth inhibition of KRAS‑ and EGFR‑mutant lung adenocarcinoma by cosuppression of STAT3 and the SRC/ARHGAP35 axis. Oncol. Rep. 2018, 40, 1761–1768.
- Aramburu, A.; Zudaire, I.; Pajares, M.J.; Agorreta, J.; Orta, A.; Lozano, M.D.; Gúrpide, A.; Gómez-Román, J.; Martinez-Climent, J.A.; Jassem, J.; et al. Combined clinical and genomic signatures for the prognosis of early stage non-small-cell lung cancer based on gene copy number alterations. BMC Genomics 2015, 16, 752.
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616.
- Garmendia, I.; Pajares, M.J.; Hermida-Prado, F.; Ajona, D.; Bértolo, C.; Sainz, C.; Lavín, A.; Remírez, A.B.; Valencia, K.; Moreno, H.; et al. YES1 Drives Lung Cancer Growth and Progression and Predicts Sensitivity to Dasatinib. Am. J. Respir. Crit. Care Med. 2019, 200, 888–899.