The third group is the “microsatellite instable” (MSI) colorectal cancer thatis caused by DNA mismatch repair gene failure, resulting in genetic hypermutability. High MSI was found in 75% of this group, which is often linked with hypermethylation and
MLH1 gene silence, whereas the remaining 25% had mutations in the mismatch-repair and polymerase (POLE) genes [
4]. Generally, genomic instability can cause aggregation of mutations in genes that are responsible for normal cell regulation and growth, such as proto-oncogenes and tumor suppressor genes [
5]. It can also derange the normal cell repair system, induce epigenetic changes in DNA, and produce non-functional proteins that could threaten the healthy cells. Notably, the significant types of genomic instability involved in the development of colorectal cancer are chromosomal instability but microsatellite stable and microsatellite instability (MSI) [
6]. Markedly, MSI is often associated with the CpG island methylator phenotype and hypermutation, which is essentially found in the right colon [
7]. Furthermore, parallel investigations revealed that the mismatch repair gene
MLH1 was hypermethylated and silenced in these MSI-positive tumors. The fact that inhibiting methylation repaired the mismatch repair deficit in colon cancer cell lines supported the hypothesis that hypermethylation causes MSI through
MLH1 silencing [
3]. MSI affects the size of the mononucleotide or dinucleotide repeats, which are also known as microsatellites, existing all over the genome. It occurs when the strand slippage within the repetitive DNA sequence element failed to be repaired. Such instability resulting from the loss of mismatch-repair function of proteins in DNA can further contribute to the inactivation of the tumor suppression pathway [
6].
A cancerous tumor can be characterized by low frequency of somatic mutations such as single nucleotide variants (SNVs), copy number aberrations (CNAs), structural variations, and indels. As indicated by the name, SNVs are aroused by a single nucleotide variant that occurred in one particular genetic position, while CNAs are the amplifications or deletions of copies of a DNA region at a larger scale. However, structural variation is used to describe an area of DNA that is 1 kb or bigger in size and can include inversions, balanced translocations, and genomic imbalances, which are also known as copy number variations. Insertions and deletions, called indels, are changes to the DNA sequence that result in the addition or deletion of one or more nucleotides [
8]. Only a small percentage of all somatic changes, known as driver mutations, offer a selective advantage to cancer cells, whereas the vast majority of somatic mutations are passenger mutations that do not contribute to the illness [
9]. Inter-tumor heterogeneity, where cancer genomes do not share a similar set of somatic mutations and most of the different metastatic tumors bear a different kind of mutation in the same patient, is the most remarkable trait of the cancer mutational landscape [
10]. Besides, in less than 5% of all patients with a specific cancer type, a small number of gene mutations are found in a large portion of tumors and mostly are affected by SNVs or CNAs [
11]. Inter-tumor heterogeneity impedes efforts to discover driver genes with driver mutations by recognizing commonly mutated genes that are mutated in a statistically high proportion of patients [
12]. The nature of the driver mutations in targeting normal functional genes, groups of interacting proteins, as well as signaling and molecular pathways, is one of the causes of inter-tumor heterogeneity [
13].
2. Driver Genes in CRC
Multistep tumorigenesis develops through the gradual collection and alterations of driver genes in colorectal cancer. Less than 1% of human genes can potentially turn into cancerous driver genes which are actively capable of controlling cell survival and fate, as well as affecting normal genome stability [
10,
20]. For a mature cell to become cancerous, it has to undergo phases of breakthrough, expansion, and invasion within 20 to 30 years, involving at least 2 to 3 driver gene mutations. It begins with the first driver mutation which minimally benefits the cell to survive and turns into a proliferating hyperplastic lesion. This could increase the risk of acquiring the second driver gene mutation and further leads to the third driver gene mutation as the cell gained autonomy and immortality, as well as the ability to self-renew. In the case when a third driver gene is involved, the tumor cell is upgraded to become invasive and metastatic. At this point, the malignant cells disseminate without the assistance of other driver mutations [
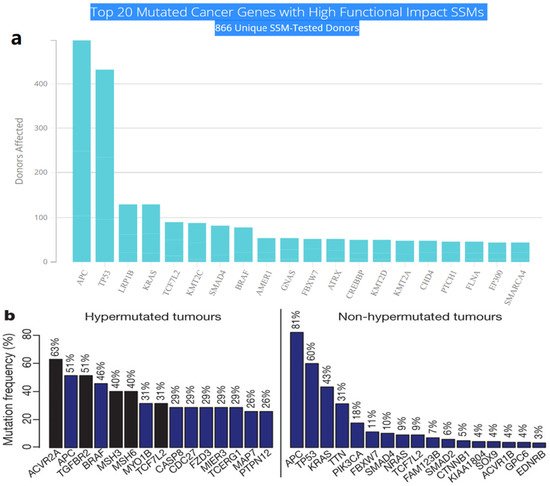
21]. The International Cancer Genome Consortium (ICGC) platform shows the top 20 mutated genes in CRC such as APC, TP53, LRP1B, KRAS, and BRAF, which are significantly impacted by single somatic mutations that also have high functional impact as shown in
Figure 1a. ICGC is a global platform that has compiled data on 670,946 unique somatic mutations and molecular profiles from 866 donors for CRC patients. These collected data are grouped into three CRC-related projects, namely, colon adenocarcinoma—TGCA, USA (COAD-US), non-Western colorectal cancer—China (COCA-CN), and rectum adenocarcinoma—USA (READ-US). In the same context, the Cancer Genome Atlas project profiled genomic changes in three cancer types; glioblastoma and ovarian carcinoma, in addition to colon and rectal cancer, among 20 different cancer types with a comprehensive molecular characterization for each one of them [
7]. In this project, 276 samples were analyzed for a genome-scale investigation of promoter methylation, exome sequence, DNA copy number, and messenger and microRNA expression. Frequent mutations were revealed in ARID1A, SOX9, and FAM123B, in addition to the expected APC, TP53,
SMAD4, PIK3CA, and KRAS mutations as shown in
Figure 1b. Furthermore, amplifications in ERBB2 and the “newly-discovered” IGF2 that might be drug-targeted were also identified in the same project, are two examples of recurrent copy-number alterations.
Figure 1. (
a) The top 20 mutated genes with high functional impact involved in colorectal cancer extracted from the ICGC Data Portal in three projects: Colon Adenocarcinoma—TCGA, US, Adenocarcinoma, non-Western (China), Rectum Adenocarcinoma—TCGA, US.
https://dcc.icgc.org/ (accessed on 15 December 2021) (
b) Significantly mutated genes in hypermutated and non-hypermutated tumors adopted from The Cancer Genome Atlas Network [
7].
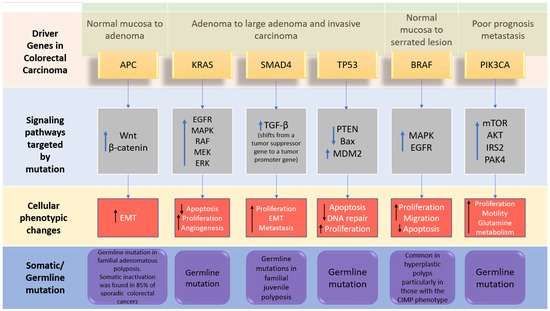
The genome-wide investigations strongly confirm the links between commonly altered driver genes and human colorectal cancer (
Figure 2). Tumorigenesis is generated in the presence of mutant driver genes such as APC, KRAS,
SMAD4, TP53, PIK3A, ARID1A, and SOX9, in intestinal epithelial cells using organoid culture systems [
7,
22]. In addition to the previously stated genes, other changed genes identified to be implicated in colorectal cancer carcinogenesis include FBXW7, BRAF, TCF7L2, PIK3CA, GNAS, CBX4, ADAMTS18, TAF1L, CSMD3, ITGB4, LRP1B, and SYNE1 [
23]. APC, KRAS, BRAF, PIK3CA,
SMAD4, and TP53 are the six CRC driver genes, with APC, KRAS, PIK3CA, and p53 being the most often altered. Mutations in APC, KRAS, and BRAF occur early in the transition phase from normal epithelium to adenoma, whereas PIK3CA mutation and loss of
SMAD4 and P53 (due to mutations or epigenetic silencing) occur late, allowing tumor cells to invade surrounding tissues and metastasize, transforming the adenoma into a carcinoma. Mutations in APC, TP53, and KRAS, as well as, to a lesser extent,
SMAD4, are related to metastatic conditions while being highly associated with MSI [
24]. The APC (adenomatous polyposis coli) gene is thought to be the gatekeeper gene for CRC, with mutations reported in 83% of all cases [
25]. KRAS contributes significantly to carcinogenesis by activating the RAF–MAPK and PI3K pathways. TGF-β signaling, on the other hand, promotes epithelial cell differentiation, acting as a tumor suppressor in colorectal cancer. Furthermore, FBXW7 is a component of the ubiquitin ligase complex, which eliminates proto-oncogene products by degradation, acting as a tumor suppressor, and Fbxw7 disruption promotes intestinal carcinogenesis. According to recent findings, mutant p53 affects gene expression globally via a gain-of-function mechanism, which promotes cancer [
22]. APC mutations frequently occur concomitantly with KRAS or TP53 mutations, or both. This triad predicts poor prognosis, whereas BRAF, ITGB4, CBX4, CSMD3, SYNE1, FBXW7, and TAF1L are substantially linked to MSI but not to metastatic illness [
20].
Figure 2. The driver genes and signaling pathways involved across the CRC adenoma–carcinoma sequence from the transition of normal epithelium through to the metastasis stage in colorectal cancer (adopted from [
6]). IRS2; insulin receptor substrate 2, MDM2; Mouse double minute 2 homolog, mTOR; Mammalian target of rapamycin. PAK4; p21 (RAC1) activated kinase 4, EMT; epithelial–mesenchymal transition.
3. Inactivation of Tumor-Suppressor Genes
3.1. Adenomatous Polyposis Coli (APC)
Apart from generating familial adenomatous polyposis (FAP), mutations in both alleles of the APC gene have a rate-limiting role in most sporadic CRC. The cascade of molecular events induced by the loss of APC function can subsequently contribute to the malignancy of the large bowel [
26]. One of the crucial intracellular components, β-catenin, which is also the binding partner of APC, is found to be involved in the Wingless/Wnt signal transduction pathway. Wnt signaling pathway, which is promoted by the mutation of gene encoding the APC protein, initiates genomic colorectal carcinogenesis. Normally, the unoccupied, phosphorylated β-catenin is attached to the destruction complex in healthy cells without being stimulated by the extracellular Wnt signal. The destruction complex consists of the scaffolding protein axin, as well as other components such as APC, conductin, and glycogen synthase kinase 3-β (GSK3β). If not attached to that complex, the nuclear localization of β-catenin proteins will create a transcription factor favoring the cellular activation of oncogenic activities. Therefore, as the APC protein complex loses its function due to its encoding gene mutation, Wnt signaling pathway is activated with increasing oncogenic β-catenin protein nuclear localization. Somatic mutations and deletion of APC encoding gene are discovered in most sporadic colorectal adenomas and carcinomas, while germ-line mutations were found in familial adenomatous polyposis [
6,
27].
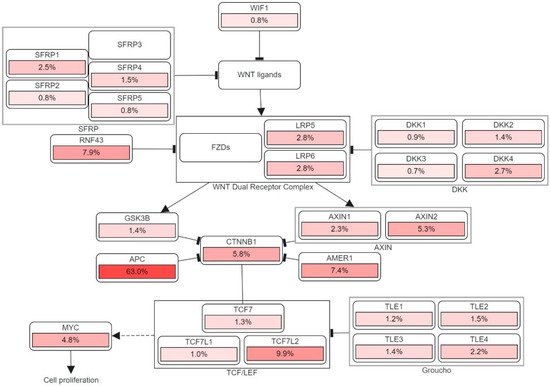
Figure 3 illustrates the detailed pathway.
Figure 3. The genetic pathways and frequencies of mutations collected from 13 studies and 4535 samples in the cBioportal platform that results in deregulation in Wnt signaling pathway, leading to the cell phenotypic modification. The dotted arrow illustrates induction. CTNNB1: Catenin Beta 1, TCF7: Transcription Factor 7, DKK: Dickkopf WNT Signaling Pathway Inhibitor, LRP: LDL Receptor Related Protein, SFRP: Secreted Frizzled Related Protein. The percentage under each gene represents the percent of mutated/altered samples related to the profiled ones in those studies [
30,
31,
32,
33,
34,
35,
36,
37,
38].
CyclinD1 and MYC are the first two discovered downstream targets in Wnt signaling pathway responsible for tumor formation due to their capabilities in cell apoptosis, proliferation, and controlling or disrupting cell-cycle progression. Direct and indirect Myc activation via the Wnt/β-catenin pathway have distinct carcinogenic effects in the intestinal epithelium [
28]. On the other hand, β-catenin overexpression in the cytoplasm, may accelerate malignant transformation in colorectal tumors by stimulating cyclin D1 expression [
29]. Other Wnt target genes, including matrilysin, CD44, and the urokinase-type plasminogen activator receptor, appear to be more involved in tumor promotion than in tumor initiation [
26].
3.2. TP53 Inactivation Pathway
Generally, the most frequent type of gene alterations that occur in human cancers are the p53 gene mutations. The transcriptional activity of the p53 protein is inactivated in most colorectal cancers by a missense mutation of the first allele and a 17p chromosomal deletion that extinguishes the second allele. The functional domains of TP53 are: transactivation domain (TAD), core domain that identifies specific DNA sequences, tetramerization domain, and the C-terminal domain that is responsible for the regulation of p53 activity [
39]. As both p53 alleles are eliminated, tumor suppression activities in its pathway were shut down and the existing large adenomas become more invasive. The activity of p53 pathway can also be suppressed by the mutation in gene encoding BAX, which normally induces cell apoptosis, in colorectal cancers with mismatch-repair defects [
40]. P53 protein is a stress-inducible transcription factor, acting as a functional regulator in a variety of downstream genes in multiple cell-signaling processes. In order to control the level of p53 from being excessive in normal cells, the negative regulator of p53 i.e., MDM2 will be upregulated to degrade p53 by regulating the ubiquination of p53. An abnormal amount of p53 can lead to cell apoptosis, cell cycle arrest or senescence triggered by DNA damage, hypoxia, and oncogene activation, as well as other cellular stresses [
41].
Two pathways are triggered simultaneously upon the activation of p53, namely, the intrinsic mitochondrial and the extrinsic death-receptor-induced apoptotic pathways. Down along the intrinsic pathway, the pro-apoptotic B-cell lymphoma-2 (Ccl-2) family proteins (i.e., BAX, Noxa and PUMA) are induced while the pro-survival Bcl-2 are downregulated instead. As the result of the permeabilization of its outer membrane, the substance cytochrome c, which is released from the mitochondria, binds to Apaf-1 and forms a complex. The complex then activates initiator caspase-9, followed by executioner capase−3, −6, and −7 [
42]. In the extrinsic pathway, the expressions of death receptors (DFs) Fas (CD95/APO-1), DR5 (TRAIL-R2), and PIDD (p53-induced protein with death domain) are upregulated as p53 is activated [
43]. Additionally, a co-transcription factor named AFT3 assists p53 in maximizing the expression of DR5, which is a trans-membrane tumor necrosis factor (TNF), in CRC induced by DNA damage. DR5 consists of a death domain which binds to the tumor necrosis factor-related apoptosis-inducing ligand (TRIAL) and activates the extrinsic apoptotic pathway that triggers cell death [
44].
A variety of small compounds have been designed to target and stabilize certain mutant versions of p53, restoring wild-type (WT)-like transcriptional activity and causing mutant tumor cells to undergo cell cycle arrest or apoptosis. The nine most common mutations of p53 protein (R175H, R248Q, R273H, R248W, R273C, R282W, G245S, R249S, Y220C) account for around 30% of all its cancer-driving mutations [
45]. PRIMA-1 and its methyl analog APR-246 are potential small molecules that interact with the DNA binding domain of mutant p53, encouraging correct folding/function and, as a result, increase the production of pro-apoptotic genes Puma, Noxa, and Bax in p53 mutant cells [
46]. The Y220C mutation is the ninth most common p53 missense mutation, that is linked to more than 100,000 new cancer cases each year. The Y220C pocket’s hydrophobic and “druggable” characteristics make it a good candidate to be targeted by small-molecule stabilizers. The mutation-induced crevice is far away from the p53 surfaces involved in DNA recognition or protein–protein interactions, allowing for creation of tailored chemical agents that stabilize the DNA binding domain without interfering with its natural substrate binding [
45]. Several powerful lead compound families that bind Y220C pockets have been identified in recent years using fragment-based and in silico screening approaches. PK9328 is a carbazole derivative that was identified by computational screening techniques fit in the p53-Y220C binding pocket with a low micromolar affinity and has a significantly decreased cell viability in various Y220C cancer cell lines [
47]. Moreover, the pyrazole derivative PK7088 restored p53-Y220C transactivation and downstream upregulation of p21 and Noxa expression, correlated with cell cycle arrest and apoptosis [
48].
3.3. TGF-β Tumor Suppressor Pathway
Because it affects cell proliferation, differentiation, apoptosis, and homeostasis, TGF-β signaling is critical in the context of inflammation and cancer. TGF signaling suppresses epithelial growth in normal tissues but promotes tumor cell proliferation in malignant tissues. This phenomenon is called the TGF-β paradox, and instead of its typical nature of inhibiting the epithelial growth in normal tissues, the activated signaling pathway stimulates tumor progression in cancerous cells [
49]. Tumor cells’ release of TGF-β also reduces the immune response to the tumor, allowing it to develop further [
50]. Two serine/threonine protein kinases (Type I and Type II receptors) and a series of downstream substrates (SMADs) are involved in TGF-β signaling. Type 2 receptors work as activators to phosphorylate type I receptors, and type 1 operate as propagators to carry the signal downstream to cytoplasmic proteins [
51]. Bone morphogenetic protein (BMP) type 1 receptors phosphorylate SMAD1/5/8 after ligand binding, whereas TGF- type I and activin type 1 receptors phosphorylate SMAD2/3. These sets of SMAD proteins are known as receptor-regulated SMAD (R-SMAD). Trimerization with a common-mediator
SMAD4 and two R-SMAD molecules, which is facilitated by the phosphorylation of two C-terminal serine R-SMAD residues, leads to its translocation into the nucleus to bind to the DNA binding site [
52]. The other non-canonical, SMAD-independent pathways that can be transduced by the TGF-β superfamily ligands include phosphoinositide 3-kinase (PI3K)/Akt, Rho/Rho-associated protein kinase (ROCK) pathways, as well as multiple types of mitogen-activated protein kinase (MAPK) [
53].
TGFBR2 mutations are frequently found in MSI-H CRC (colorectal cancer with microsatellite instability-high frequency). Mismatch repair genes are silently expressed in MSI-H CRC cells due to germline mutations in genes such as MutL homolog 1 (
MLH1), MutS homolog 2 (
MSH2),
MSH6, and Postmeiotic segregation increased 2 (
PMS2), or
MLH1 promoter hypermethylation. The genes that are affected by the germline mutations are usually MutL homolog 1 (
MLH1), MutS homolog 2 (
MSH2), MutS homolog 6 (
MSH6), Postmeiotic segregation increased 2 (
PMS2) or
MLH1 promoter hypermethylation. TGFBR2 mutations, which are often discovered in MSI-H CRC, have the ability to convert normal epithelial cells into malignant ones in the colon [
54]. Therefore, the malignant phenotype of the affected CRC cells will arise via Hippo, MAPK, and Wnt-β-catenin signaling pathways [
55]. The second type of TGF-β Signaling in CRC is the mutation and deletion of the suppressor gene
SMAD4 as a key transcription factor in this pathway. Many genes in the 18q21 chromosomal region are frequently affected by the loss of heterozygosity including
SMAD2 and
SMAD4 may contribute to forming microsatellite-stable CRC. Because it is a transcription factor for TGF-β signaling, the loss of tumor suppressor gene
SMAD4 impairs canonical TGF-β signaling [
7]. The non-canonical TGF-signaling route is the third signaling pathway. Although
SMAD4 deletion inhibits canonical TGF-β signaling, it modifies BMP signaling via a non-canonical route to enhance CRC metastasis via activation of the Rho/ROCK pathway, resulting in EMT, migration, and invasion.
SMAD4 deficiency also activates alternate MEK/ERK pathways, promoting cell death, migration, and invasion [
56].